Experimental design considerations and statistical analyses in preclinical tumor growth
inhibition studies - Python tutorial
Author
Sébastien Wieckowski
Published
January 3, 2025
The goal of this project is to create a comprehensive and accessible Python-based resource for researchers
conducting preclinical cancer studies using animal models. It’s an adaptation of the R tutorial provided by Bonato and colleagues to
emphasize Python’s strengths in data analysis, visualization, and machine learning, offering a modern toolkit for
scientists.
1
Introduction
Cancer, a complex disease driven by the relentless evolution of somatic cells, presents one of
the most formidable challenges in modern medicine. This evolution, marked by the accumulation of genetic
alterations and the selection of advantageous subclones within a tumor, fundamentally shapes the disease’s
progression and response to treatment. Understanding this intricate process is not merely a biological
curiosity; it is a cornerstone of effective drug development.
Pre-clinical animal models, serving as invaluable surrogates for human tumors, offer a crucial
platform to dissect this evolutionary process and assess the therapeutic potential of novel compounds. By
replicating key aspects of tumor growth, heterogeneity, and treatment response, these models enable researchers
to identify promising drug targets, optimize dosing regimens, identify biomarkers, evaluate compound safety, and
predict clinical outcomes. However, extracting meaningful insights from these studies requires rigorous
statistical analysis to quantify tumor growth inhibition, assess drug synergy, and discern subtle
differences between treatment groups.
This chapter explores the critical intersection of animal models, tumor evolution, and statistical analysis in
the quest for effective cancer therapies. We delve into the nuances of study design, endpoint selection, and
data analysis techniques tailored for tumor growth inhibition (TGI) studies. Our aim is to
equip researchers with the knowledge and tools necessary to navigate the complexities of pre-clinical data,
ultimately accelerating the translation of promising compounds from the laboratory bench to the patient’s
bedside.
1.1 Objectives
This tutorial aims to equip researchers with the essential tools for designing and analyzing preclinical
animal studies focused on evaluating the efficacy of anticancer drugs. The primary objective is to guide
readers in modeling tumor growth dynamics and quantifying tumor growth inhibition (TGI) effects induced by
novel compounds compared to control treatments (e.g., placebo or standard therapies).
By the end of this tutorial, we will:
Understand the fundamental principles of study design for preclinical cancer research, including
considerations for animal model selection, treatment allocation, and data collection strategies.
Gain proficiency in statistical methods for analyzing tumor growth data, both in terms of continuous
measures (tumor volume, growth rate) and time-to-event outcomes (survival, tumor progression).
Interpret and draw conclusions from TGI analyses, assessing the statistical significance and clinical
relevance of treatment effects.
Apply these techniques to real-world data using simulated examples and Python code snippets provided
throughout the tutorial.
While no prior experience with Python or R programming is required, a basic understanding of statistical
principles (e.g., hypothesis testing, P values) and familiarity with concepts like linear regression, survival
analysis, and non-parametric methods will greatly enhance the learning experience. My open-source Jupyter Book,
providing a Python-based companion to the “Intuitive Biostatistics” book, might be a valuable resource for
this work. We encourage to actively engage with this Jupyter Notebook, adapting them to own datasets to
solidify the understanding!
2 Data preparation
2.1 Toy sample generation
Let’s first prepare a function that will generate a simulated dataset for a group of animals
with specific characteristics.
Code
import numpy as npimport pandas as pdfrom scipy.stats import t as t_dist # Avoid mixing t value with t-distributiondef simulate_exp_growth( group_name: str, duration: int, initial_tumor_volume: float, group_growth_rate_day_pct: float, growth_rate_cv: float, residual_cv: float, n_animals: int, max_tumor_volume: float, df=None) -> pd.DataFrame:""" Simulates tumor growth data for a group of animals over a given duration. Args: group_name (str): Name of the group (e.g., "control", "therapy_a"). duration (int): Duration of the study in days. initial_tumor_volume (float): Initial tumor volume (mm^3). group_growth_rate_day_pct (float): Average daily growth rate of the group (%). growth_rate_cv (float): Coefficient of variation for the growth rate within the group. residual_cv (float): Coefficient of variation for the measurement error. n_animals (int): Number of animals in the group. max_tumor_volume (float): Maximum allowed tumor volume before removal (mm^3). df (int, optional): Degrees of freedom for the t-distribution. If None, defaults to n_animals - 1. Returns: pandas.DataFrame: Simulated data with columns: day, animal, group, tv (Tumor Volume). """# Initialize an empty list to store DataFrames for each animal in the group group_data = []# Loop over each animal in the groupfor i inrange(n_animals):# Create an array of time points (days) when measurements are taken, starting from# day 0 up to the end of the study (`duration`). Measurements are taken every 3 days. days = np.arange(0, duration +1, 3)# Simulate individual growth rates## individual_growth_rate = np.random.normal(# group_growth_rate_day_pct / 100,# group_growth_rate_day_pct * growth_rate_cv / 100,# len(days) - 1# )## Since we're simulating tumor growth data, and the sample size is usually small# (below 15 animals per group), using the t distribution could be a reasonable choice.# It will introduce slightly more variability into the simulated data, reflecting the# uncertainty associated with small samples and the stochastic nature of tumor growth.# Shift and scale the t-distribution for individual growth rate df = n_animals -1if df isNoneelse df # Default df if not provided individual_growth_rate = t_dist.rvs( df=df, loc=group_growth_rate_day_pct /100, scale=group_growth_rate_day_pct * growth_rate_cv /100, size=len(days) -1 )# Note: if the growth rates are strictly positive and tend to be right-skewed (some# tumors grow much faster than others), the gamma distribution can be a good option.# Initialize an empty array to store the tumor volumes over time# ln_tumor_volume = np.empty(len(days))# we're now working with log-normal tumor volumes directly (instead of log-transformed values) tumor_volume = np.empty(len(days))# Simulate the initial log-transformed tumor volume## If the initial tumor sizes are tightly controlled and have low variability,# a normal distribution might be suitable. Log-normal or truncated normal# distributions could also be considered. tumor_volume[0] = np.random.normal( loc=initial_tumor_volume, scale=initial_tumor_volume * residual_cv )# Iterate over time points (days) to simulate tumor growthfor t inrange(1, len(days)):# ln_tumor_volume[t] = np.random.normal(# ln_tumor_volume[t - 1] + individual_growth_rate[t - 1] * days[t],# np.sqrt(np.log(residual_cv**2 + 1))# )## the log-normal distribution is a popular choice for modeling tumor volumes# over time, as it captures the right-skewed nature of tumor growth data.## Calculate mean and standard deviation of the log-normal distribution# Apply exponential growth to the previous tumor volume (not log-transformed) mean = tumor_volume[t-1] *\ np.exp(individual_growth_rate[t-1] * days[t])# Calculate the mean and standard deviation of the underlying normal# distribution for log-normal mean_log = np.log(mean)# With std_log = sqrt(ln(CV² + 1)) std_log = np.sqrt(np.log(residual_cv**2+1))# Simulate tumor volume at time t directly from the log-normal distribution# The `np.random.lognormal` function takes the mean and standard deviation# of the underlying normal distribution as input. Remember, if a variable X# follows a log-normal distribution, then the logarithm of that variable,# ln(X), follows a normal distribution. tumor_volume[t] = np.random.lognormal( mean=mean_log, sigma=std_log, )# Considere Gompertz Growth Model that describes sigmoidal growth patterns,# which are often observed in tumors.# # Transform log-tumor volumes back to the original scale (tumor volumes in mm^3).# tumor_volume = np.exp(ln_tumor_volume)# Find indices where tumor volume exceeds the maximum allowed. welfare_missing = np.where(tumor_volume > max_tumor_volume)[0]iflen(welfare_missing) >1:# Exclude from its first occurrence until the next timepoint for survival . tumor_volume[welfare_missing[1:]] = np.nan# Create a DataFrame for the current animal's data animal_data = pd.DataFrame( {"day": days,"animal": f"{group_name}_{i +1}","group": group_name,"tv": tumor_volume, } )# Add the animal's data to the list of group data group_data.append(animal_data)# Concatenate all animal dataframes into one for the entire group# ignore_index=True resets the index of the resulting DataFrame.return pd.concat(group_data, ignore_index=True)
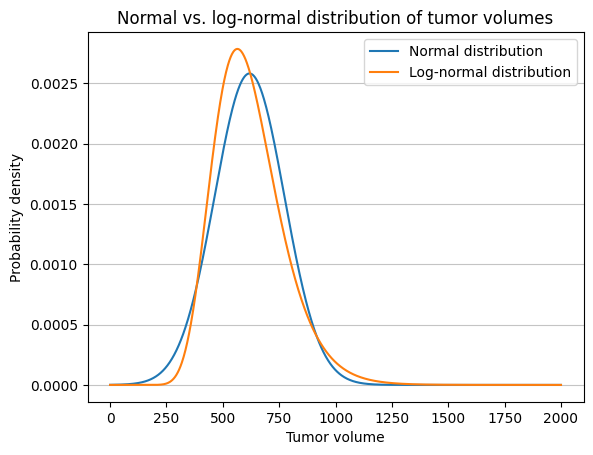
The normal
distribution is symmetric and centered around its mean. The standard deviation we calculated directly
affects the spread of the distribution on both sides of the mean. The log-normal distribution is
right-skewed and has a long tail on the right side. The std_log calculated affects the
spread of the underlying normal distribution of the log-transformed values. When exponentiated to get back to
the original scale, this leads to a different pattern of variability compared to the normal distribution, the
peak of the log-normal distribution being lower and shifted to the left compared to the peak of the normal
distribution.
By using the initial_tumor_volume as the geometric
mean and the residual_cv to derive the geometric standard deviation, you’re
working with parameters that are directly meaningful in the context of the log-normal
distribution. This ensures that the simulations are based on the correct understanding of the
geometric nature of tumor growth data.
Code
import matplotlib.pyplot as pltfrom scipy.stats import norm, lognorm# Initiating values for tumor_volume and residual_cvtumor_volume =600residual_cv =0.25# Calculate parameters for the underlying normal distribution of the log-normalmean_log = np.log(tumor_volume)std_log = np.sqrt(np.log(residual_cv**2+1))# Calculate the mean and SD of the equivalent normal distributionmean_normal = np.exp(mean_log + std_log**2/2)std_normal = np.sqrt( (np.exp(std_log**2) -1)* np.exp(2*mean_log + std_log**2))# When we use std = initial_tumor_volume * residual_cv, we're calculating the standard deviation# directly from the initial tumor volume and the coefficient of variation. This is correct if# residual_cv is the CV of the normal distribution. However, in the original simulation setup,# residual_cv represents the coefficient of variation of the log-normal distribution,# not the normal distribution.# Create x values for plottingx = np.linspace(0, 2000, 1000)# Calculate PDF values for normal and log-normal distributionsnormal_pdf = norm.pdf(x, loc=mean_normal, scale=std_normal)lognormal_pdf = lognorm.pdf(x, s=std_log, scale=np.exp(mean_log))# Plot the PDFsplt.plot(x, normal_pdf, label='Normal distribution')plt.plot(x, lognormal_pdf, label='Log-normal distribution')plt.xlabel('Tumor volume')# plt.xscale("log")plt.ylabel('Probability density')plt.title('Normal vs. log-normal distribution of tumor volumes')plt.legend()plt.grid(axis='y', alpha=0.75)
To demonstrate the statistical analysis techniques discussed in this chapter, we’ll utilize a simulated
dataset generated using the following parameters and assumptions:
Experimental groups:
One control group (Vehicle).
Two single-agent therapy groups (Therapy_A, Therapy_B).
One combination therapy group (Therapy_Combo_AB).
Sample size: each group starts with 15 animals (n_animals = 15).
Study duration: the study lasts for 18 days (duration = 18).
Initial tumor volume: all animals start with a tumor volume around 100 mm3
(initial.tumor.volume = 100).
Measurement frequency: tumor volumes are measured every 3 days.
Measurement error: the coefficient of variation (CV) for measurement error is 25%
(residual.cv = 0.25).
Tumor growth rates: average daily tumor growth rates (in %) are:
Vehicle: 5%
Therapy_A: 4.5%
Therapy_B: 3.5%
Therapy_Combo_AB: 1.3%
Growth rate variability: the coefficient of variation for growth rates within each group is 10%
(growth.rate.cv = 0.1).
Maximum tumor volume: animals whose tumor volumes reach 2000 mm3 are removed from the study
(max.tumor.volume = 2000).
Code
# Set a seed for reproducibilitynp.random.seed(444)# Simulate the vehicle (control) group datavehicle_group = simulate_exp_growth( group_name="control", duration=18, initial_tumor_volume=100, group_growth_rate_day_pct=5, growth_rate_cv=0.1, residual_cv=0.25, n_animals=15, max_tumor_volume=2000)# Simulate data for the single-agent therapy A grouptherapy_a_group = simulate_exp_growth( group_name="therapy_a", duration=18, initial_tumor_volume=100, group_growth_rate_day_pct=4, growth_rate_cv=0.1, residual_cv=0.25, n_animals=15, max_tumor_volume=2000)# Simulate data for the single-agent therapy B grouptherapy_b_group = simulate_exp_growth( group_name="therapy_b", duration=18, initial_tumor_volume=100, group_growth_rate_day_pct=3, growth_rate_cv=0.1, residual_cv=0.25, n_animals=15, max_tumor_volume=2000)# Simulate data for the combination therapy A+B grouptherapy_ab_group = simulate_exp_growth( group_name="therapy_combo_ab", duration=18, initial_tumor_volume=100, group_growth_rate_day_pct=1.3, growth_rate_cv=0.1, residual_cv=0.25, n_animals=15, max_tumor_volume=2000)# Combine all group data into a single DataFrameTGI_dataset_test = pd.concat( [ vehicle_group, therapy_a_group, therapy_b_group, therapy_ab_group ], ignore_index=True)# Let's have a look at the dataset structure and valuesprint(TGI_dataset_test.head())
day animal group tv
0 0 control_1 control 103.815784
1 3 control_1 control 128.940403
2 6 control_1 control 176.041978
3 9 control_1 control 209.166393
4 12 control_1 control 442.556814
For comparison with the R tutorial we can import the dataset TGI.dataset from the original
study.
Code
TGI_dataset = pd.read_csv("TGI_dataset.csv")TGI_dataset.columns = [col.lower() for col in TGI_dataset.columns]for col in TGI_dataset.select_dtypes(include='object').columns: TGI_dataset[col] = TGI_dataset[col].astype(str).str.lower()print(TGI_dataset.head(5))
day animal group tv
0 0 control_1 control 104.825993
1 3 control_1 control 125.137266
2 6 control_1 control 155.847247
3 9 control_1 control 238.687772
4 12 control_1 control 335.119128
2.2 Analyzing tumor growth dynamics
2.2.1 Key statistics with a custom function
To gain deeper insights into the tumor growth patterns within each experimental group, we’ll utilize a
function to calculate summary statistics for tumor growth inhibition (TGI). This will allow us to compare
the average tumor volumes across groups at different time points and assess the variability within each
group.
Code
import warningswarnings.simplefilter(action='ignore', category=FutureWarning)warnings.simplefilter(action='ignore', category=DeprecationWarning)def summary_stats_tgi(dataset: pd.DataFrame) -> pd.DataFrame:"""Calculates geometric mean, standard error, and 95% confidence intervals for tumor volumes per group per day. Args: dataset (pd.DataFrame): TGI dataset with columns 'day', 'group', and 'tv' (tumor volume). Returns: pd.DataFrame: Summary statistics with columns 'day', 'group', 'est' (geometric mean), 'se' (standard error), 'lb95_est' (lower 95% CI bound), and 'ub95_est' (upper 95% CI bound). """# Split the dataset by group and day grouped_data = dataset.groupby(['group', 'day'])# Apply the summary statistics calculation to each group summary_list = []for (group, day), group_df in grouped_data:# First, we take the natural logarithm of the tumor volumes# Drop missing values (NaN) log_tumor_volumes = np.log(group_df['tv'].dropna())# Analyze the log-transformed values using standard normal distribution techniques# To get back to the original scale (tumor volumes), we exponentiate the mean and confidence# interval limits. Since the exponential is the inverse of the logarithm, this back-transformation# gives us the geometric mean and the corresponding confidence interval on the original scale. geometric_mean = np.exp(np.mean(log_tumor_volumes))# In R, the default behavior of the var() function is to use a delta degrees of freedom (ddof) of 1.# This means it calculates the sample variance, which is considered an unbiased estimator of the# population variance. NumPy's np.var function, by default, uses ddof = 0 (population variance),# but not pandas's var which used default ddof = 1. se = np.sqrt(log_tumor_volumes.var() /len(log_tumor_volumes)) lb95 = geometric_mean * np.exp(norm.ppf(0.025) * se) ub95 = geometric_mean * np.exp(norm.ppf(0.975) * se) summary_list.append({"day": day,"group": group,"est": geometric_mean,"se": se,"lb95_est": lb95,"ub95_est": ub95 })# Combine results into a DataFramereturn pd.DataFrame(summary_list)
While we’re simulating tumor volumes from a log-normal distribution, we actually use the normal
distribution to calculate the mean and confidence intervals. The log-normal distribution is inherently
defined as a distribution whose logarithm follows a normal distribution. This means if a variable X is
log-normally distributed, then \(\log(X)\) follows a normal distribution.
When we want to calculate summary statistics like the mean and confidence interval for a log-normal
distribution, it’s often easier and more reliable to work with the logarithms of the data rather than the
raw data itself. Indeed, the mean of the log-transformed data directly corresponds to the location parameter
of the log-normal distribution. Also, the log-transformed data follows a normal distribution, which is
symmetric. This symmetry makes it easier to calculate confidence intervals and apply standard statistical
tests.
While it’s theoretically possible to calculate summary statistics directly on the log-normal distribution,
it’s often more complex and prone to errors. The log-normal distribution is skewed, so its arithmetic mean
might not be a good representation of the central tendency. Additionally, calculating confidence intervals
directly on the log-normal distribution can be computationally challenging.
Leveraging Pandas’ groupby and describe functions, we can effortlessly generate a
detailed statistical profile of the tumor volumes within each experimental group, including the number of
animals remaining, at each time point.
Code
TGI_dataset.groupby(['group', 'day']).describe()
tv
count
mean
std
min
25%
50%
75%
max
group
day
control
0
15.0
102.337373
12.879400
73.966753
98.079914
102.927819
108.765041
125.245267
3
15.0
135.822689
32.528511
83.542341
113.084926
127.282145
159.104481
200.072588
6
15.0
189.766983
47.882196
120.659156
159.487281
183.848327
218.303025
279.768096
9
15.0
301.584275
113.275488
120.438583
236.362885
282.724282
359.417484
563.845041
12
15.0
530.467881
247.383477
205.588181
333.563703
456.265546
707.509967
970.984050
15
15.0
1347.140701
670.145035
296.057302
870.395873
1286.366006
1795.646514
2831.206338
18
15.0
3420.889383
2090.362199
463.809975
2017.002897
3228.041028
4630.789148
8345.411255
therapy_a
0
15.0
106.963038
28.635230
58.913855
91.820106
97.169935
113.085365
160.933938
3
15.0
113.424589
45.696562
47.949666
84.560273
99.675973
131.490110
218.594997
6
15.0
147.484120
57.812170
45.955902
118.466128
131.192080
187.781208
270.608753
9
15.0
217.481620
99.489732
62.945368
155.360546
175.624492
272.227813
393.041179
12
15.0
356.223597
178.620840
84.066759
240.887216
350.549399
423.646575
798.510798
15
15.0
616.542902
287.643446
173.321385
412.393238
609.756321
768.338336
1300.752659
18
15.0
1272.715957
616.831030
491.715154
830.020666
939.946099
1727.996591
2449.440952
therapy_b
0
15.0
101.622632
27.748183
68.683737
86.996987
94.462453
100.606739
173.368783
3
15.0
116.502890
45.676643
51.471512
77.642927
111.360294
151.265746
190.062664
6
15.0
159.943658
66.658128
71.677215
112.450580
152.966335
201.741230
285.400361
9
15.0
223.773995
105.409381
71.919811
148.479905
211.324123
275.358286
430.777369
12
15.0
320.154044
177.458595
85.357512
224.919608
283.700964
421.497751
704.788052
15
15.0
559.365932
385.954352
115.675295
370.667577
443.944166
689.627500
1523.108747
18
15.0
847.034379
613.333316
183.144309
508.833347
762.757727
939.605722
2468.363846
therapy_combo_ab
0
15.0
98.863617
21.913776
58.542840
80.452217
103.228822
112.582789
131.965342
3
15.0
102.033695
34.218192
56.524456
81.264018
97.960340
112.704304
188.781830
6
15.0
111.866578
58.485060
36.540022
75.760590
101.452698
128.681882
260.732431
9
15.0
130.357569
70.031341
37.381473
74.024599
127.813497
172.560568
274.715958
12
15.0
167.975680
109.856143
34.714108
84.479192
149.857299
208.723595
413.430485
15
15.0
213.252799
123.938002
40.303314
115.875287
212.318566
265.859798
489.926716
18
15.0
287.799543
203.230603
63.786859
150.290977
234.453907
333.979129
783.215058
The pivot_table function in Pandas is a powerful tool for generating custom summary tables,
allowing us to easily compare geometric means of tumor volumes or other relevant metrics across different
experimental conditions.
Code
from scipy.stats import gmeanprint("Geometric mean:")TGI_dataset.pivot_table( values='tv', index='day', columns='group', aggfunc=gmean)
Geometric mean:
group
control
therapy_a
therapy_b
therapy_combo_ab
day
0
101.536731
103.594835
98.736314
96.364433
3
132.261775
105.617074
108.043137
97.289897
6
184.300154
136.011443
147.498501
99.155402
9
281.463182
195.629951
199.556878
111.844686
12
477.111360
312.601850
270.856421
136.879392
15
1169.973866
551.161945
443.544059
178.214612
18
2750.176428
1137.049884
677.167458
225.100813
Code
print("Arithmetic mean:")print( TGI_dataset.pivot_table( values='tv', index='day', columns='group',#aggfunc='mean' # Default function ))
print("Standard error of the geometric mean:")TGI_dataset.pivot_table( values='tv', index='day', columns='group', aggfunc=lambda x: np.sqrt(np.log(x).var()/len(x)) # standard error)
Standard error of the geometric mean:
group
control
therapy_a
therapy_b
therapy_combo_ab
day
0
0.034077
0.067338
0.061430
0.062295
3
0.061715
0.100664
0.105557
0.081205
6
0.064576
0.113316
0.108302
0.132551
9
0.102086
0.127840
0.133412
0.154870
12
0.125217
0.145064
0.164438
0.176595
15
0.153067
0.132813
0.191394
0.170720
18
0.195191
0.128526
0.182431
0.195905
Finally we can put all the statistics together using grouby and custom aggregate functions,
including the use of t-distribution for computing the confidence interval.
Code
def se(x):# Returns the standard error of the log-transformed datareturn np.sqrt(np.log(x).var() /len(x))def ci95(x):# Returns the 95% confidence interval of the geometric mean (t-distribution) log_mean = np.log(gmean(x)) # Mean of the log-transformed data sem = se(x) # Standard error of the log-transformed data t = t_dist.ppf(.975, df=len(x)-1) # Critical t-value with q=1-α/2# Calculate the confidence interval in the log scale and then exponentiatereturn [round(np.exp(bound), 4) for bound in (log_mean-t*sem, log_mean+t*sem)]print(TGI_dataset.groupby(['group', 'day'])['tv'].agg([gmean, se, ci95]))
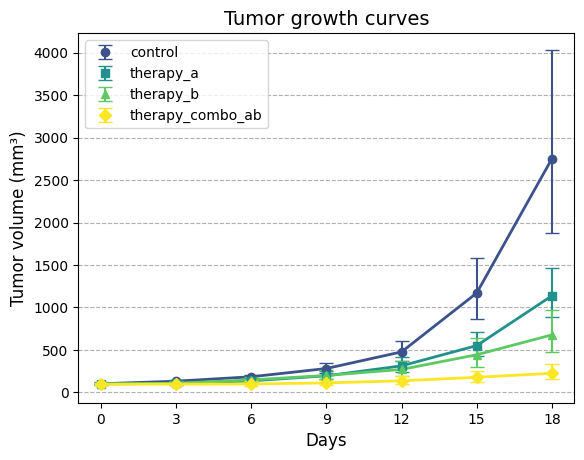
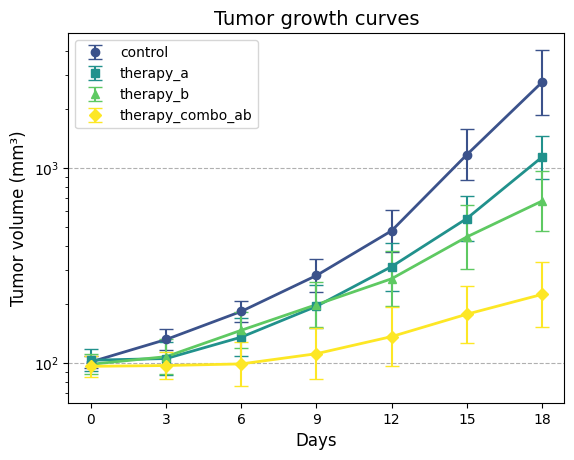
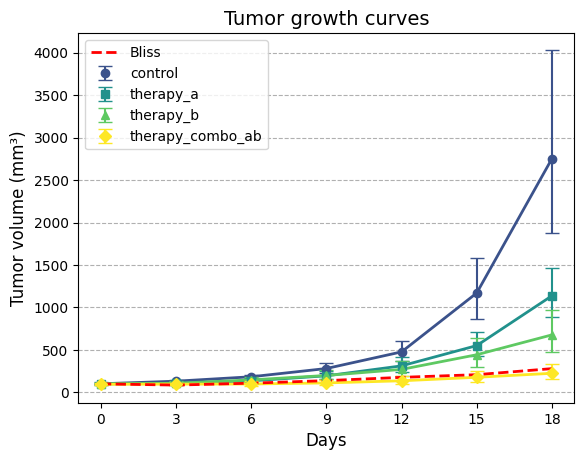
To effectively visualize and compare the tumor growth patterns among the experimental groups, we’ll create
a plotting function that can display tumor growth curves with their associated confidence intervals. This
will allow us to assess the impact of different therapies on tumor growth over time, both in the original
scale and on the logarithmic scale.
Code
def plot_tgi_stats( summ_stats: pd.DataFrame, ln_scale: bool=False, colormap: str="viridis") ->None:"""Plots tumor growth curves with 95% confidence intervals. Args: summ_stats (pd.DataFrame): Dataframe with summary statistics (output of `summary_stats_tgi`). ln_scale (bool, optional): If True, plot on natural log scale. Defaults to False. colormap (str, optional): Colormap to use for group colors. Defaults to "viridis". """# Map groups to colors and symbols groups = summ_stats["group"].unique() n_groups =len(groups)# Map groups to colors from the colormap, and markers colors = plt.get_cmap(colormap)(np.linspace(0.25, 1, n_groups))# Different markers for each group markers = ['o', 's', '^', 'D', '*', 'v', 'x', 'p']# Set up plot fig, ax = plt.subplots()# Plot each groupfor i, group inenumerate(groups): group_data = summ_stats[summ_stats["group"] == group] ax.errorbar( group_data["day"], group_data["est"], yerr=[ group_data["est"] - group_data["lb95_est"], group_data["ub95_est"] - group_data["est"]], fmt=markers[i], color=colors[i], label=group, capsize=5) ax.plot( group_data["day"], group_data["est"], color=colors[i], linewidth=2)# Formatting ax.set_xlabel("Days", fontsize=12)# Ensure x-axis ticks are integers ax.set_xticks(group_data['day'].unique()) ax.set_xticklabels(group_data['day'].unique(), fontsize=10)if ln_scale: ax.set_yscale("log") ax.set_ylabel("Tumor volume (mm³)", fontsize=12) ax.set_title("Tumor growth curves", fontsize=14) ax.grid(which='major', axis='y', linestyle='--') ax.legend(loc='upper left', fontsize=10)
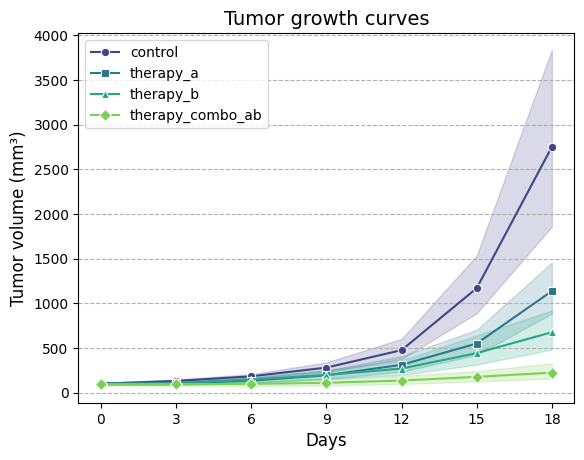
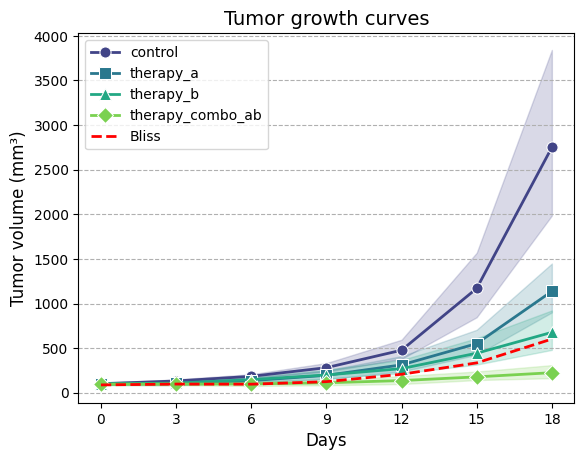
Seaborn is a powerful data visualization
library built on top of Matplotlib. It offers a higher-level interface for creating informative and
attractive statistical graphics, often with less code than Matplotlib. The code is much
more concise and does not require a separate function to calculate summary statistics. It also handles the
calculation of geometric means and confidence
intervals internally (using bootstrapping method).
Code
import seaborn as sns# Create line plot of tumor volume over time for each group, using geometric meanax = sns.lineplot(# Input DataFrame containing tumor growth data, not the summary statistics data=TGI_dataset, x='day', # Column name for time points (x-axis) y='tv', # Column name for tumor volumes (y-axis)# Column name for grouping variable (different lines/colors for each group) hue='group',# Column name for assigning different line styles to groups (automatic) style='group',# Disable automatic dashes for different line styles (we'll use markers) dashes=False, markers=["o", "s", "^", "D"], # Manually assign markers for each group estimator=gmean, # Use geometric mean as the estimator for central tendency# Show 95% confidence intervals as error bars errorbar=('ci', 95), palette='viridis', # Color palette to use for different groups# Show confidence intervals as filled bands around the lines err_style='band')# Optionally plot on a logarithmic scale# plt.yscale('log')# Ensure x-axis ticks are integersax.set_xticks(TGI_dataset['day'].unique())ax.set_xticklabels(TGI_dataset['day'].unique())ax.set_xlabel("Days", fontsize=12)ax.set_ylabel("Tumor volume (mm³)", fontsize=12)ax.set_title("Tumor growth curves", fontsize=14)ax.grid(which='major', axis='y', linestyle='--')ax.legend(loc='upper left', fontsize=10);
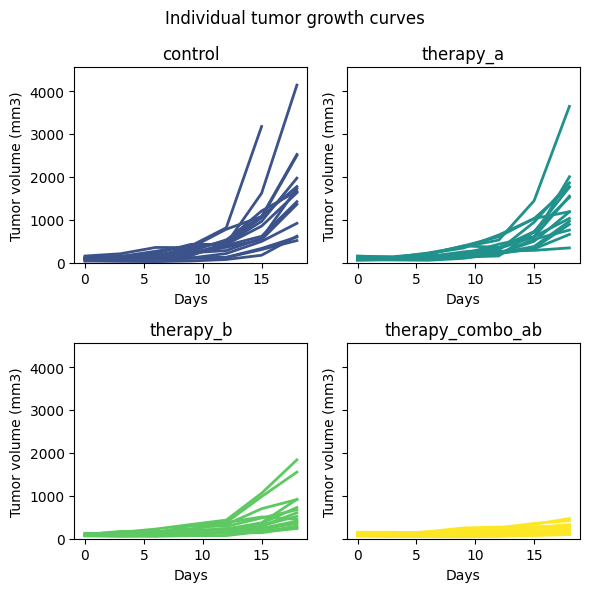
While summary statistics like the geometric mean and confidence intervals provide valuable insights into
the average tumor growth trends, visualizing individual tumor growth curves offers a deeper understanding of
the underlying variability and heterogeneity within each group. It allows us to identify outliers, observe
patterns of response to treatment, and assess the overall distribution of tumor growth trajectories.
To facilitate this visualization, we’ll create a function plot_individual_curves that
automatically generates subplots, one for each group, displaying the individual growth curves of all animals
within the group. This approach simplifies the plotting process, making it easier to explore the data and
compare treatment effects across groups.
By examining these individual curves alongside the summary statistics, we can gain a more comprehensive
picture of the tumor growth dynamics in our preclinical study. This holistic view can be crucial in
interpreting the results, identifying potential issues with experimental design or data collection, and
making informed decisions about the next steps in the drug development process.
Code
def plot_individual_curves( dataset: pd.DataFrame, colormap: str="viridis", max_cols: int=2) ->None:"""Plots individual tumor growth curves for each animal, with one subplot per group. Args: dataset (pd.DataFrame): DataFrame with columns 'day', 'tv' (tumor volume), 'group', and 'animal'. colormap (str, optional): Colormap to use for group colors. Defaults to "viridis". max_cols (int, optional): Maximum number of columns in the plot grid. Defaults to 2. """ groups = dataset["group"].unique() n_groups =len(groups)# Map groups to colors from the colormap colors = plt.get_cmap(colormap)(np.linspace(0.25, 1, n_groups))# Calculate number of rows and columns for subplots num_cols =min(n_groups, max_cols) num_rows = (n_groups + num_cols -1) // num_cols# Find the maximum tumor volume across all groups max_tumor_volume = dataset['tv'].max()# Create the figure and axes fig, axes = plt.subplots( nrows=num_rows, ncols=num_cols,# Adjust figsize based on num_cols figsize=(3* num_cols, 3* num_rows), sharey=True, )# Flatten the axes array for easier iteration axes = axes.ravel()# Plot individual curves for each groupfor i, group inenumerate(groups): ax = axes[i] group_data = dataset[dataset["group"] == group]for animal in group_data["animal"].unique(): animal_data = group_data[group_data["animal"] == animal] ax.plot( animal_data["day"], animal_data["tv"], color=colors[i], label=animal, lw=2)# Formatting ax.set_title(group) ax.set_xlabel("Days") ax.set_ylabel("Tumor volume (mm3)") ax.set_ylim(0, max_tumor_volume *1.1) # Set y-axis limits# Hide any unused subplots if the number of groups is not evenfor j inrange(n_groups, num_cols * num_rows): axes[j].set_visible(False)# Add overall title and adjust layout plt.suptitle("Individual tumor growth curves") plt.tight_layout() plt.show()
Code
# Plot the current TGI_datasetplot_individual_curves(TGI_dataset_test)
3 Experimental design
3.1 Overview of animal models in TGI studies
Preclinical TGI studies rely on various animal models, each with strengths and limitations that influence
their suitability for different research questions. While no model perfectly replicates human tumors, careful
model selection can improve research efficiency and reduce the risk of late-stage drug development failures.
Subcutaneous: easy to establish, monitor tumor growth with calipers.
Orthotopic: more accurately mimics tumor microenvironment, allows for studying invasion and
metastasis.
Limitations (orthotopic): technically challenging, tumor measurement may require sacrifice or imaging.
Genetically engineered mouse models (GEMMs):
Advantages: recapitulate syngeneic tumor-host relationship, can study multiple endpoints (tumor
growth, metastasis).
Limitations: asynchronous tumor development, long development time, difficult to measure deeply
located tumors.
Organoid models:
Advantages: preserve tumor heterogeneity and microenvironment, standardized, controlled setting.
Limitations: still under development, utility in drug discovery needs further investigation.
The choice of animal model depends on the specific research question, budget, and available resources. While
cell line xenografts offer high throughput and standardization, PDX models provide valuable insights into
personalized treatment responses. Orthotopic models offer a more realistic tumor microenvironment, while GEMMs
allow for studying tumor-host interactions and multiple endpoints. Organoids hold promise for future research,
but their applications are still being explored.
3.2 Endpoints in preclinical TGI studies
Preclinical TGI studies assess how well a new therapy (\(T\)) inhibits tumor
growth compared to a control group (\(C\)). Researchers have various options
for choosing endpoints, which are the measures used to evaluate treatment effects.
Tumor growth inhibition (TGI): this is a quantitative measure of how much a therapy
reduces tumor size compared to the control. It’s often expressed as a percentage (\(\text{TGI}\%\)), where 0% indicates no inhibition and 100% indicates complete
inhibition, or when the tumor completely stops growing or disappears: \[\text{TGI}_\text{T|C} = 1 - \frac{f(Y_T)}{f(Y_c)}\] where \(f\) is a function whose output depends on \(Y_T\) (treated group) and \(Y_C\) (control
group), based directly on tumor sizes or volumes, or derived from tumor volume measurements.
Derived endpoints from tumor volumes
Absolute tumor volume: the actual (average) size of the tumor at a given time point, i.e.,
\(f(Y) = Y\).
Change in tumor volume (∆TGI): the difference in tumor volume between the start and
end of treatment, e.g., \(f(Y) = Y_1 - Y_0\).
Area under the curve (AUC): the total area under the tumor growth curve over the study period.
Tumor growth rate: the rate at which the tumor grows over time.
Time-to-event (TTE) endpoints: these focus on the time it takes for a specific event to occur, such as
tumor reaching a certain size, metastasis or animal death (although using death as an endpoint is
discouraged for ethical reasons). TTE can be useful for seeing long-term treatment impact.
Categorical endpoints: This is a more qualitative way of looking at results. Instead of exact numbers, we
put responses into categories like “complete response” (CR), “partial response” (PR), etc.
This is similar to how results are often discussed in human cancer trials. The function \(f\) in TTE can be conceptualized as comparing relative risk
estimates between groups.
3.3 Quantifying treatment effects
Having generated our simulated dataset and explored the individual tumor growth trajectories, we now need a
way to quantify the effectiveness of the treatments in a standardized manner. For this purpose, we’ll
calculate two key metrics:
Tumor Growth Inhibition (TGI): this metric directly assesses the reduction in tumor size in the treated
group compared to the control group at specific time points. A higher TGI indicates a more effective
treatment.
TGI-Delta (∆TGI): this metric accounts for baseline tumor size differences between groups, providing a
more nuanced view of treatment efficacy. It measures the change in tumor volume from a defined baseline time
point, allowing for a comparison of growth changes rather than absolute sizes.
The following function, calculate_tgi_from_data, allows us to calculate either TGI or TGI-delta
from our simulated dataset.
Code
def calculate_tgi_from_data( dataset: pd.DataFrame,type: str='TGI', time_points: list=None, baseline: int=0, treatment_name: str=None, control_name: str="control") -> pd.DataFrame:"""Calculates either TGI or TGI_delta from a tumor growth dataset. Args: dataset (pd.DataFrame): Dataframe with columns 'day', 'group', and 'tv' (Tumor Volume). type (str, optional): Types of TGI to calculate ('TGI' or 'TGI_delta'). Defaults to "TGI". time_points (list, optional): List of days at which to calculate TGI. If None, calculates for all days. baseline (int, optional): Baseline day for TGI_delta calculation. Required if 'TGI_delta' is in `type`. treatment_name (str, optional): Name of the treatment group. Defaults to None (calculates for all groups vs. control). control_name (str, optional): Name of the vehicle (control) group. Defaults to "control". Returns: pd.DataFrame: DataFrame containing TGI or TGI_delta results. Raises: ValueError: If no treatment_name provided and no other group than control found in the dataset """# Ensure treatment_name is specifiedifnot treatment_name: # not None returns True non_control_groups = dataset[dataset['group']!= control_name]['group'].unique()iflen(non_control_groups) ==0:raiseValueError("No treatment groups found in the dataset.")# otherwise we take the first non-control group from the list as the treatment group treatment_name = non_control_groups[0]# Add baseline for calculating TGI_delta time_points = [baseline] +\ time_points iftype=="TGI_delta"else time_points# Filter control and treatment data control_data = dataset[(dataset["group"] == control_name) & ( dataset["day"].isin(time_points))] treatment_data = dataset[(dataset["group"] == treatment_name) & ( dataset["day"].isin(time_points))]# Calculate means control_means = control_data.groupby("day")["tv"].mean().reset_index() treatment_means = treatment_data.groupby("day")["tv"].mean().reset_index()# Calculate TGI or TGI_deltaiftype=="TGI": result_data = control_means.merge( treatment_means, on="day", suffixes=("_control", "_treatment")) result_data["tgi_pct"] = (1- (result_data["tv_treatment"] / result_data["tv_control"])) *100 result_data["contrast"] =f"{treatment_name} vs. {control_name}"return result_data[["contrast", "day", "tgi_pct"]]eliftype=="TGI_delta": control_baseline = control_data[control_data["day"] == baseline] treatment_baseline = treatment_data[treatment_data["day"] == baseline]# Calculate mean baseline tumor volumes for both groups control_baseline_mean = control_baseline["tv"].mean() treatment_baseline_mean = treatment_baseline["tv"].mean()# Adjust tumor volumes relative to their respective baseline means control_means["tv_control_adjusted"] = control_means["tv"] -\ control_baseline_mean treatment_means["tv_treatment_adjusted"] = treatment_means["tv"] -\ treatment_baseline_mean# Merge and calculate TGI_delta result_data = control_means.merge(treatment_means, on="day") result_data["tgi_delta_pct"] = (1- (result_data["tv_treatment_adjusted"] / result_data["tv_control_adjusted"])) *100 result_data["contrast"] =f"{treatment_name} vs. {control_name}"# mask to exclude baseline from the report ix = result_data['day'] == baselinereturn result_data.loc[~ix, ["contrast", "day", "tgi_delta_pct"]]else:raiseValueError("Invalid type. Please specify either 'TGI' or 'TGI_delta'.")
Code
# Example 1 - Single time point TGI. Imagine we want to assess the efficacy of# therapy_b compared to the control group at day 18 of the study.calculate_tgi_from_data( dataset=TGI_dataset,type='TGI', time_points=[18, 21, 24, 27], # it doesn't hurt if days don't exist treatment_name="therapy_b", control_name='control')
contrast
day
tgi_pct
0
therapy_b vs. control
18
75.239352
Code
# Example 2 - TGI-delta over multiple time points. If we're interested in# how the effect of therapy_B changes over time, we can calculate TGI-delta# at multiple time points relative to a baseline (e.g., day 0).calculate_tgi_from_data( dataset=TGI_dataset,type='TGI_delta', time_points=[6, 12, 18],# baseline=0, # default treatment_name="therapy_b",# control_name='control' # default name of the control group)
contrast
day
tgi_delta_pct
1
therapy_b vs. control
6
33.293736
2
therapy_b vs. control
12
48.956823
3
therapy_b vs. control
18
77.538042
Why choose one over the other?
TGI: use when the baseline tumor volumes are relatively similar between the treated and control
groups. This provides a straightforward measure of tumor reduction at specific time points.
TGI-Delta: use when there are notable differences in baseline tumor volumes. TGI-delta accounts
for this initial variation, focusing on the change in tumor growth relative to the starting point.
3.4 Enhanced data analysis with Pandas
The calculate_tgi_from_data custom function prioritizes the arithmetic mean for TGI calculations
due to its focus on relative changes, potential use in linear models, simplicity, and consistency with
baseline adjustment. However, the geometric mean remains relevant for understanding the tumor volume data
itself and might be preferred in other analysis contexts.
Pandas provides powerful tools, such as groupby, agg and transform,
for direct DataFrame manipulation, enabling efficient calculations like:
Mean tumor volume: computing both arithmetic and geometric means for all treatment and control groups at
each time point.
TGI calculation (geometric mean): determining TGI for each treatment group at each time point, using the
geometric means for a more accurate representation of the central tendency in log-normally distributed data.
ΔTGI calculation (arithmetic mean): calculating ∆TGI for all treatment groups and time points, utilizing
arithmetic means to account for differences in baseline tumor sizes, similarly to the function above.
This streamlined approach eliminates the need for additional functions and prepares the data for more
advanced statistical analysis.
Code
# we apply arithmetic 'mean' and scipy gmean to tumor volumes# grouped by treatment group and dayTGI_dataset.groupby(['group', 'day'])['tv'].agg(['mean', gmean])#TGI_dataset.groupby(['group', 'day']).agg({'tv': ['mean', gmean]})
mean
gmean
group
day
control
0
102.337373
101.536731
3
135.822689
132.261775
6
189.766983
184.300154
9
301.584275
281.463182
12
530.467881
477.111360
15
1347.140701
1169.973866
18
3420.889383
2750.176428
therapy_a
0
106.963038
103.594835
3
113.424589
105.617074
6
147.484120
136.011443
9
217.481620
195.629951
12
356.223597
312.601850
15
616.542902
551.161945
18
1272.715957
1137.049884
therapy_b
0
101.622632
98.736314
3
116.502890
108.043137
6
159.943658
147.498501
9
223.773995
199.556878
12
320.154044
270.856421
15
559.365932
443.544059
18
847.034379
677.167458
therapy_combo_ab
0
98.863617
96.364433
3
102.033695
97.289897
6
111.866578
99.155402
9
130.357569
111.844686
12
167.975680
136.879392
15
213.252799
178.214612
18
287.799543
225.100813
Code
# TGI at multiple time-points# use .xs to access values from each group in the hierarchical index(TGI_dataset.groupby(['group', 'day']).tv # equivalent to ['tv'].mean().transform(lambda x: 1- x / x.xs('control', level=0)).unstack(level=0) # Pull out the outer-most index and push it up into columns#.loc[[12, 15], ['therapy_a', 'therapy_b']])
group
control
therapy_a
therapy_b
therapy_combo_ab
day
0
0.0
-0.045200
0.006984
0.033944
3
0.0
0.164907
0.142243
0.248773
6
0.0
0.222815
0.157158
0.410506
9
0.0
0.278869
0.258005
0.567757
12
0.0
0.328473
0.396469
0.683344
15
0.0
0.542332
0.584775
0.841700
18
0.0
0.627958
0.752394
0.915870
Code
# Adjusted geometric means of the tumor volume( TGI_dataset .groupby(['group', 'day']) .agg(adjusted_geometric_mean=('tv', gmean)) # named aggregation .transform(lambda x: x - x.xs(0, level=1)) .unstack(level='group'))
adjusted_geometric_mean
group
control
therapy_a
therapy_b
therapy_combo_ab
day
0
0.000000
0.000000
0.000000
0.000000
3
30.725044
2.022239
9.306823
0.925464
6
82.763423
32.416607
48.762187
2.790968
9
179.926451
92.035116
100.820564
15.480252
12
375.574628
209.007015
172.120106
40.514959
15
1068.437135
447.567110
344.807745
81.850178
18
2648.639697
1033.455049
578.431143
128.736380
Code
# TGI-delta (arithmetic mean) over all groups and time points( TGI_dataset .groupby(['group', 'day']) ['tv'] .agg('mean') .transform(lambda x: x - x.xs(0, level=1)) .transform(lambda x: 1- x/x.xs('control', level=0)) .unstack(0) # Pull out the outer-most index and push it up into columns .drop(index=0, columns='control') # Remove 'control' and NaN cells)#.loc[[6, 12, 18], ['therapy_b']]
group
therapy_a
therapy_b
therapy_combo_ab
day
3
0.807033
0.555618
0.905329
6
0.536529
0.332937
0.851275
9
0.445318
0.386935
0.841935
12
0.417793
0.489568
0.838572
15
0.590634
0.632277
0.908107
18
0.648716
0.775380
0.943067
3.5 Log-transformed tumor volumes and geometric mean
In the previous analysis, we used original (untransformed) tv values of tumor volume and the
arithmetic meean for different reasons:
Focus on relative change: TGI and ΔTGI are measures of the relative change in tumor volume
compared to a control or baseline. These metrics are often expressed as percentages or ratios, which are
inherently multiplicative in nature. Using the original (untransformed) tumor volumes for TGI calculations
might be more directly interpretable in terms of these relative changes.
Baseline adjustment: TGI_delta involves subtracting the baseline tumor volume. Using the original scale
for this subtraction maintains consistency and avoids potential issues with interpreting differences on the
log scale.
Simplicity and convention: in some cases, using the original scale for TGI might be preferred for its
simplicity and wider understandability, especially when communicating results to a broader audience.
We will see in the next sections that the log-transformed log_tv values are used in ANOVA and
group comparisons, which can be supported by this rationale:
Normality assumption: ANOVA and many statistical tests assume that the data is normally distributed. Tumor
volume data often follows a log-normal distribution, meaning the logarithms of the tumor volumes
are normally distributed. By using log_tv, we meet the normality assumption of these tests,
making the results more reliable.
Variance stabilization: log transformation can help stabilize the variance across different groups,
especially if the variance increases with the mean tumor volume. This is important for the validity of ANOVA
and other tests that assume equal variances.
Linearity: in some cases, the relationship between the predictors (e.g., treatment group, time) and the
outcome (tumor volume) might be more linear on the log scale. Using log_tv can improve the fit
of linear models and make the interpretation of the coefficients more straightforward.
3.6 Measuring tumor volume in preclinical studies
Accurately measuring tumor volume in animal studies is essential for evaluating how well a treatment works,
as tumor size is linked to the number of active cancer cells. Different methods exist, each with its own
advantages and drawbacks:
Ex vivo weighing: this involves surgically removing the tumor and weighing it, offering precise volume
measurements. However, it’s limited to a single time point at the end of the study.
In vivo caliper measurements: this non-invasive method is quick and suitable for easily accessible tumors
(e.g., those implanted under the skin). It involves measuring tumor dimensions (length, width, and height)
and calculating volume using geometric formulas. Tumor volume is often calculated assuming an ellipsoid
shape (\(Y = 1/6 \pi \times L \times W \times H\)), where \(L\) is the length, \(W\) the width and \(H\) the height, or a simplified approximation for equal height and width
(\(Y = 1/2 \times L \times W^2\)).
Imaging techniques (MRI, CT, Ultrasound): for deeply located tumors, imaging methods provide detailed
internal views and precise measurements, even for complex shapes. However, these techniques can be costly
and raise ethical concerns due to the need for anesthesia and potential stress on animals.
Choosing the right measurement method depends on factors like tumor type, location, desired accuracy, study
duration, and ethical considerations.
3.7 Understanding and mitigating measurement errors in TGI studies
Measurement errors, both systematic (bias) and random, are inherent to any technique used to measure tumor
size in animal studies. These errors can significantly affect the accuracy, precision, and validity of
preclinical research findings.
Types of measurement errors:
Bias (systematic errors): these are consistent deviations from the true tumor size, often caused by
factors such as instrument limitations, observer variability, or assumptions about tumor shape.
Caliper-based measurements, while convenient and cost-effective, are particularly prone to bias due to
factors like tissue interference and subjective interpretation.
Random errors: these are unpredictable fluctuations in measurements that affect the precision of the
results. While unavoidable, their magnitude can be reduced through experience, training,
and standardized procedures.
Impact of measurement errors:
Bias: if not addressed, bias can lead to inaccurate conclusions about the effectiveness
of a treatment. For example, a biased measurement tool might consistently overestimate tumor size in one
group, leading to an incorrect conclusion about treatment efficacy.
Precision: random errors reduce the precision of measurements, making it harder to detect
statistically significant differences between treatment groups. This can result in inconclusive results or
underpowered studies.
Strategies for mitigating measurement errors:
Study design: randomization, blinding, and stratification can help minimize bias and
ensure balanced groups.
Measurement techniques: choose the most appropriate technique based on the tumor type, location, and
desired accuracy. Imaging techniques (MRI, CT, ultrasound) are generally more precise but can be costly and
raise animal welfare concerns.
Standardization and training: consistent use of instruments, standardized protocols, proper quality
control, and extensive training can reduce both bias and random error.
Additional considerations:
Small tumor volumes: measurement accuracy tends to decrease for small tumors due to equipment
limitations and the challenges of measuring irregular or inflamed tissues.
Noise in data: even with careful design, TGI studies can produce noisy data. Sample size
calculations are crucial to ensure sufficient statistical power to detect treatment effects despite this
noise.
By understanding and addressing measurement errors, researchers can improve the reliability and validity of
their preclinical studies, leading to more informed decisions about promising cancer therapies.
3.8 Handling discrepant measurements and missing data in TGI studies
In preclinical TGI studies, unexpected or missing data points can occur. It’s essential to have a clear plan
for dealing with these issues to maintain the validity and integrity of the study’s conclusions.
Discrepant measurements (outliers):
When to exclude: remove outliers only if there’s a clear understanding of a specific error during the
study (e.g., incorrect dosage, tumor cell engraftment issue). These exclusion criteria
should be defined in advance.
When to include: if there’s no obvious error, it’s best to include all animals to avoid introducing bias.
Non-parametric tests can be helpful as they’re less sensitive to outliers.
Missing Data:
Random missingness:
MCAR (missing completely at random): no systematic difference between animals with missing and
complete data.
MAR (missing at random): missingness is explainable by a known factor unrelated to treatment or
response (e.g., cage-specific illness).
Informative censoring: missingness is more likely in certain groups. Two common scenarios:
Large tumors: animals are sacrificed due to welfare concerns (usually >2000 mm³). This can lead to
underestimation of tumor volume in untreated groups.
Small tumors: highly effective treatments shrink tumors below the detection limit (<50 mm³). This
can lead to bias and imprecision in estimates.
Handling informative censoring:
Limited analysis: one option is to analyze TGI only up to the time point when the first animal was removed
due to tumor size.
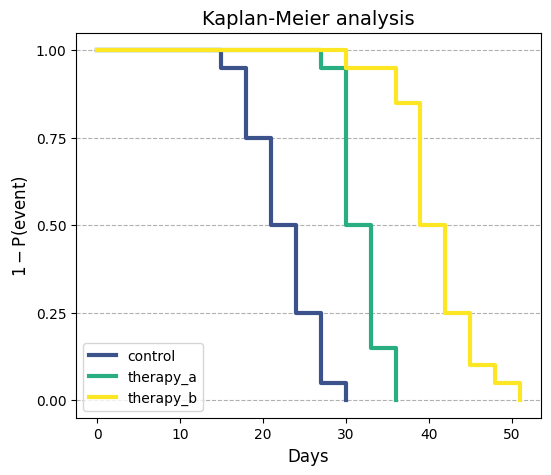
Time-to-event analysis: another approach is to use time-to-event analysis, where the “event” is the tumor
reaching a large size. This eliminates the issue of informative censoring related to large tumor sizes.
Key points:
Handling missing data and outliers requires careful consideration to avoid introducing bias.
Pre-specifying criteria for data exclusion is crucial for maintaining objectivity.
Different approaches are suitable depending on the type of missingness or outlier.
Time-to-event analysis can be a valuable tool when dealing with informative censoring due to large tumor
volumes.
4 Statistical analysis in preclinical cancer studies
After measuring tumor volumes in animal studies, the standard method to assess the impact of anticancer
treatments is to determine whether there are statistically significant differences between
treatment and control groups. The choice of statistical test depends on several factors:
Study endpoint: whether the endpoint is continuous (tumor volume, growth rate), time-to-event (tumor
progression, death), or categorical (tumor response categories).
Number of time points: whether the study involves a single time point assessment or multiple measurements
over time.
Parametric vs. non-parametric: whether the data meet the assumptions of parametric tests (e.g.,
normality) or require non-parametric methods.
The appropriate statistical test will vary based on these factors. For example, t-tests or ANOVA might be used
for continuous endpoints at a single time point, while repeated measures ANOVA or mixed models might be used for
longitudinal data:
mindmap
root((Data))
**Categorical** - _Compare frequencies_
(**Chi-squared tests** - _Trend is not important_)
(**Cochran-Armitage** - _Trend is important_)
**Time-to-event** - _Modeling events_
(**Log-rank tests** - _non-parametric_)
(**Cox PH** models - _semi-parametric w/wo adjustment for covariates_)
**Continuous** - _Typical TGI_
[Single time-point - _usu. last day of study_]
(**T-tests** or **ANOVAs** - _w/wo adjustment for covariates_)
(**Bliss** or **HSA** - _analysis of **synergy_**)
[Multiple time-points - _several assessments throughout study_]
(**Growth rates** or **AUCs** - _summarizes into a few parameters_)
(**T-tests**, **ANOVAs**, or **ANCOVAs** - w/wo covariates, assessment at each time poin_t_)
(**Bliss** or **HSA** - _analysis of **synergy**_)
4.1 Analyzing continuous tumor volume data at a single time point
In TGI studies where tumor volume is measured only once (usually at the end of the study), the data can be
analyzed as follows:
Data structure: the tumor volume data (\(Y\)) is represented as \(y_{ij}\), where:
\(i\) is the index for each individual animal (\(i = 1, \dots, N\)) where \(N\) is the
total number of animals.
\(j\) is the index for the treatment group (\(j = 1, \dots, J\)) where \(J\) is the
number of groups.
Log transformation: tumor volumes are often log-transformed due to their right-skewed distribution. \[\stackrel{*}{y_{ij}} = \ln(y)\] Log-transforming tumor volumes in TGI
studies offers several advantages:
Normality: log-transformation helps normalize the data, making it suitable for parametric statistical
tests that assume a normal distribution.
Equal variances: it can also help stabilize the variance across different groups,
making them more comparable.
Reduced degrees of freedom: avoids using up degrees of freedom for testing normality and equal
variances after transformations.
Interpretable differences: transforms differences between groups into fractions, eliminating
counter-intuitive negative growth rates.
Enables parametric methods: allows for using standard parametric tests like t-tests (for two groups,
\(J = 2\)) or ANOVA (for multiple groups, \(J
\ge 2\)) or linear regression models, which are often more powerful than their non-parametric
counterparts.
Handling zeros: if some tumor volumes are zero, they can be replaced with the smallest measurable tumor
volume to avoid issues with log transformation, especially when there are only a few zero measurements.
This approach allows researchers to apply standard statistical tests (e.g., t-tests, ANOVA) to compare the
average log-transformed tumor volumes between treatment groups, and then back-transform the results to the
original volume scale.
4.1.1 How it works
In TGI studies with a single time point and continuous tumor volume data, a simple linear regression model
or ANOVA can be used for statistical analysis.
Log transformation: tumor volumes \(Y\) are log-transformed \(\ln(Y)\) to meet the normality assumption of the model.
Model setup: the core idea behind this model is that the log-transformed tumor volume of each animal
(\(i\)) is influenced by the treatment group (\(j\)) the animal belongs to.
The model assumes that each treatment group has a potentially different average effect on the tumor
volume, and this effect is captured by the parameters \(\alpha_j\). The
mathematical equation that describes the model is \(\stackrel{*}{y_{i}} =
\sum_{j=1}^J{\alpha_j I_{ij} + \epsilon_i}\), where \(I_{ij} =
1\) indicates the treatment \(j\) to which each animal \(i\) was assigned to, otherwise \(I_{ij} =
0\), i.e., the animal \(i\) is not in group \(j\) if \(I_{ij} = 0\), and \(\epsilon_i\) is the residual error term for animal \(i\), representing the variation in tumor volume that is not explained by
the treatment group. The model assumes these errors are normally distributed with mean 0 and variance
\(\sigma^2\), i.e., \(\epsilon_i \sim N(0,
\sigma^2)\).
For example, let’s consider two groups, control (\(j = 1\)), and
treatment A (\(j = 2\)). For an animal in the control group, \(I_{i1} = 1, I_{i2} = 0\), the equation becomes \(\stackrel{*}{y_i} = \alpha_1 + \epsilon_i\). For an animal in treatment
A, the equation becomes \(\stackrel{*}{y_i} = \alpha_2 + \epsilon_i\).
The model parameters represent:
\(\alpha_1\): the mean log tumor volume in the control group
(intercept). It’s the value we would expect if the animal received no treatment.
\(\alpha_2, \alpha_3, \dots, \alpha_J\): the mean differences in
log tumor volume between each treatment group and the control group (treatment effects).
Parameter estimation: ordinary least squares (OLS) is used to estimate the model parameters.
Hypothesis testing: statistical tests like score tests or Wald tests can be used to assess the
significance of the treatment effects.
One-sided tests: used to test if a treatment reduces tumor volume compared to the control (H1: \(\alpha_j \lt 0\)).
Two-sided tests: used to compare the tumor reduction efficacy between different treatments (H1:
\(\alpha_2 \ne \alpha_3\)).
Essentially, the model allows us to estimate the average effect of each treatment on the log tumor volume,
taking into account the natural variability between animals. By fitting this model to the data, we can then
perform statistical tests to see if the treatment effects are significantly different from zero (i.e., if
the treatments are actually working).
4.1.2 Model-based estimation of the TGI
Let’s now incorporate the predicted/estimated average tumor volume in the treatment group (\(j = T\)) and in the control group (\(j =
1\)) based on the linear model, with \(\widehat{Y_T} =
e^{\widehat{\alpha_1} + \widehat{\alpha_T}}\) and \(\widehat{Y_1} =
e^{\widehat{\alpha_1}}\), where \(\widehat{\alpha_1}\) is the
estimated mean of the log-transformed tumor volumes in the control group (intercept of the linear model),
and \(\widehat{\alpha_T}\) is the estimated difference in mean log tumor
volume between the treatment group (T) and the control group, i.e., it quantifies the treatment effect on
the log scale.
The model works with log-transformed tumor volumes. However, to calculate TGI, we need to go back to the
original scale (the actual tumor volumes). To do this, we exponentiate the log-transformed values.
As discussed earlier, the TGI is defined as the percentage reduction in tumor volume in the treatment group
compared to the control group. Mathematically, this is \(\text{TGI}_\text{T|1} = 1
- \frac{f(Y_T)}{f(Y_1)}\). In the model, we estimate the parameters \(\alpha_T\) and \(\alpha_1\) using ordinary
least squares (OLS). We denote these estimates as \(\widehat{\alpha_T}\)
and \(\widehat{\alpha_1}\). Substituting these estimates into the TGI
formula, we get \(\widehat{\text{TGI}_\text{T|1}} = 1 -
\frac{e^{\widehat{\alpha_1} + \widehat{\alpha_T}}}{e^{\widehat{\alpha_1}}}\). Using the property
that \(e^{a+b} = e^a \times e^b\), we can simplify:
By using the model-based estimates (\(\widehat{\alpha_1}\) and \(\widehat{\alpha_T}\)), you’re taking into account all the data points from
both groups and adjusting for other factors that might influence tumor growth (e.g., baseline differences
between animals). This approach can provide more accurate and reliable TGI estimates than simply comparing
the raw average tumor volumes.
Note that this method assumes that the linear model is a good fit for the log-transformed tumor volume
data.
4.1.3 Practical example
Suppose our linear model estimates are \(\widehat{\alpha_1} = 4.6\), i.e.,
the mean log tumor volume in the control group, and \(\widehat{\alpha_T} =
-0.5\), i.e., the treatment effect for group T indicating a reduction in tumor volume compared to
control. Then the estimated TGI would be \(\widehat{\text{TGI}_{T|1}} = 1 -
\exp(-0.5) \approx 0.393\). This means that the treatment is estimated to reduce the average tumor
volume by approximately 39.3% compared to the control group at the end of the study.
4.1.4 TGI analysis for single time-point data in Python
In scenarios where tumor volume measurements are taken at a single time point (typically at the end of the
study), we can employ a linear model to compare the treatment group(s) to the control group. This analysis
allows us to determine if there are statistically significant differences in tumor growth inhibition (TGI)
between the groups.
The following function, tgi_single_time, implements this analysis. It offers flexibility to either adjust
for baseline tumor volumes or analyze the raw tumor volumes directly. Additionally, it allows for both
one-sided (testing for tumor reduction) and two-sided (testing for differences in efficacy) hypothesis
tests.
Code
from typing import Listimport statsmodels.formula.api as smfdef tgi_single_time( dataset: pd.DataFrame, time_point: int, treatments_numerator: List[str], treatment_denominator: str='control', baseline_adjusted: bool=False, baseline: int=0, p_tails: int=1, correction_method: str='none',) -> pd.DataFrame:"""Estimates Tumor Growth Inhibition (TGI) at a single time point using a linear model. Args: dataset (pd.DataFrame): DataFrame with columns 'day', 'group', and 'tv' (tumor volume). time_point (int): Day at which to calculate TGI. treatments_numerator (List[str]): List of treatment group names (numerators in TGI calculation). treatment_denominator (str): Name of the comparison group (denominator in TGI calculation). Defaults to 'control'. baseline_adjusted (bool, optional): If True, adjust for baseline tumor volume. Defaults to False. baseline (int, optional): Baseline day for adjustment. Defaults to 0. p_tails (int, optional): Number of tails for the hypothesis test (1 for one-sided, 2 for two-sided). Defaults to 1. correction_method (str, optional): Method for multiple comparisons correction ('none', 'bonferroni', 'sidak'). Defaults to 'none'. Returns: pd.DataFrame: DataFrame containing TGI estimate, confidence intervals, and P value. """# Filter data for the relevant time points and groups tv_data = dataset[ (dataset['day'].isin([baseline, time_point]))& (dataset['group'].isin(treatments_numerator + [treatment_denominator])) ].copy()# Log-transform tumor volume (TV) and add lnTV column tv_data['log_tv'] = np.log(tv_data['tv'])# Baseline adjustment (if applicable)if baseline_adjusted: tv_data['log_tv_baseline'] = ( tv_data .groupby('animal') # we pick for each animal .log_tv # the log_tv value .transform('first') # of the first day, i.e., day 0 )# We filter the time point we want to analyze tv_data = tv_data[tv_data['day'] == time_point]# Build and fit the linear modelif baseline_adjusted: model = smf.ols( formula=f"log_tv ~ log_tv_baseline + C(group, Treatment(reference='{ treatment_denominator}'))", data=tv_data)else: model = smf.ols( formula=f"log_tv ~ C(group, Treatment(reference='{ treatment_denominator}'))", data=tv_data) est = model.fit() degrees_of_freedom = est.df_resid# Extract model estimates and calculate TGI# print(est.summary2()) results = est.summary2().tables[1] # returns a true DataFrame# Create a list to store the final results for all treatments model_estimates_list = []for treatment in treatments_numerator:# Extract TGI, CI, and P value for the current treatment ix =f"C(group, Treatment(reference='{ treatment_denominator}'))[T.{treatment}]" tgi = (1- np.exp(results.loc[ix, "Coef."]))# Calculate confidence intervals and P value std_err = results.loc[ix, "Std.Err."]if p_tails ==1: lb95_tgi = np.nan ub95_tgi = (1- np.exp(results.loc[ix, "Coef."] + t_dist.ppf(0.95, degrees_of_freedom) * std_err)) pvalue = t_dist.cdf(results.loc[ix, "t"], degrees_of_freedom)else: lb95_tgi = (1- np.exp(results.loc[ix, "Coef."] + t_dist.ppf(0.025, degrees_of_freedom) * std_err)) ub95_tgi = (1- np.exp(results.loc[ix, "Coef."] + t_dist.ppf(0.975, degrees_of_freedom) * std_err)) pvalue =2* (1- t_dist.cdf(abs(results.loc[ix, "t"]), degrees_of_freedom))# Adjust p-values if multiple comparison correction is specifiedif correction_method =='bonferroni': pvalue *=len(treatments_numerator)elif correction_method =='sidak': pvalue =1- (1- pvalue) **len(treatments_numerator)# Create a dataframe of the results for the current treatment model_estimates = pd.DataFrame({"contrast": [f"{treatment} vs. {treatment_denominator}"],"day": [time_point],"tgi": [tgi],"lb95_tgi": [lb95_tgi],"ub95_tgi": [ub95_tgi],"pvalue": [pvalue],"df": [degrees_of_freedom],"baseline_adj": [baseline_adjusted],"correction": [correction_method], })# Append results for this treatment to the list model_estimates_list.append(model_estimates)# Combine results into a single DataFramereturn pd.concat(model_estimates_list, ignore_index=True)
Now that we have our tgi_single_time function defined, let’s explore how it can be used to
answer different questions about the efficacy of our simulated cancer therapies.
4.1.4.1 Example 1: baseline-adjusted TGI of ‘therapy_a’ and
‘therapy_b’ at day 18
Tumors often vary in size at the start of a study due to biological or experimental factors. Adjusting
for baseline tumor volume helps control for this initial variability, making the comparison between
treatment and control groups fairer and more focused on the treatment effect itself.
This call assesses the tumor growth inhibition of ‘therapy_a’ and ‘therapy_b’ compared to the ‘control’
group at day 18, taking into account the baseline tumor volumes (day 0). We use
baseline_adjusted=True because we want to control for any potential differences in initial
tumor sizes between the two groups. This focuses the analysis on the effect of the treatment itself, not
just the final tumor size. We specify baseline=0 to indicate that the baseline measurements
were taken on day 0 of the study, though this is the default function parameter. Similarly, we specify the
treatment denominator group treatment_denominator='control' although this is the default
control group name.
4.1.4.2 Example 2: unadjusted TGI of ‘therapy_a’ and ‘therapy_b’ at
day 18
When there are substantial differences in baseline tumor sizes, not adjusting for them can lead to biased
estimates of the treatment effect. By including baseline as a covariate, we can obtain more accurate
estimates of how much the treatment actually influences tumor growth. If we’re unsure whether to adjust
for baseline, we can perform a sensitivity analysis by fitting the model both with and without the
baseline adjustment and compare the results. If the results are similar, then adjusting might not be
necessary.
Similar to example 1, this call calculates the TGI of ‘therapy_a’ and ‘therapy_b’ at day 18. However, it
doesn’t adjust for the baseline tumor volumes. We might choose not to adjust for the baseline if we are
confident that the initial tumor sizes were very similar between the groups or if we want to see the raw
effect of the treatment without considering baseline differences. Here we can see that the TGI values are
comparable in both cases, and that the P values don’t change the conclusion, sugesting that baseline
adjustment may not be necessary.
Note that the difference in degrees of freedom between baseline-adjusted and unadjusted models is
expected and statistically valid. It reflects the fact that we’re estimating an additional parameter when
adjusting for baseline, which consumes one degree of freedom from the data. Generally, a lower
‘df_residual’ leads to slightly wider confidence intervals and potentially less significant P-values, as
the model has less flexibility to fit the data. While adjusting for a significant covariate can increase
power, adjusting for a non-significant covariate can actually decrease power. This is because we’re using
up a degree of freedom without gaining much explanatory power in return.
Ideally, the decision of whether to adjust for baseline should be made before the study begins and
documented in the study protocol. This helps avoid bias and ensures the analysis plan is consistent with
the study design.
4.1.4.3 Example 3: two-sided unadjusted TGI comparison of
‘therapy_combo_ab’ vs. ‘therapy_b’ at day 12
This call compares the efficacy of the combination therapy (therapy_combo_ab) to therapy_b at day 12. We
use p_tails=2 for a two-sided hypothesis test because we’re interested in whether the
combination therapy is either better or worse than therapy_b, not just whether it’s better. We explicitly
set treatment_denominator='therapy_B' to indicate that we’re comparing the combination
therapy to therapy_b, not the control group. In this example, we don’t adjust for baseline since we’re
directly comparing two treatment groups, not a treatment to the control. We’re assuming that any baseline
differences would affect both groups similarly.
4.1.5 The problem with multiple pairwise t-tests (j > 2)
Imagine you have three treatment groups (A, B, and C) and want to compare them all. If you perform multiple
pairwise t-tests (A vs. B, A vs. C, B vs. C), you
encounter several issues:
Inflated type I error rate: each individual t-test has a chance of making a type I error (false
positive), typically set at 5%. When you conduct multiple tests, the overall probability of making at
least one false positive increases substantially. This means you’re more likely to conclude that a
treatment difference exists when it actually doesn’t.
Inefficient variance estimation: each t-test only uses the data from the two groups being compared to
estimate the variance. This ignores the information from the other groups, potentially leading to less
accurate and less stable variance estimates.
A simple linear model (or ANOVA in this case) with pairwise contrasts addresses these problems:
Controls for type I error: by performing a single overall test (e.g., F-test in ANOVA), you control the
family-wise error rate, which is the probability of making at least one type I error across all
comparisons. This ensures a more stringent threshold for declaring statistical significance.
Pooled
variance estimation: the linear model estimates the variance \(\sigma^2\) by pooling information from all the groups. This leads to a more
reliable and efficient estimate, especially when sample sizes are small.
Pairwise comparisons with adjustment: after the overall test indicates a significant difference, you can
perform pairwise comparisons using contrasts. These contrasts can be adjusted for multiple comparisons
(e.g., using Tukey’s HSD), further controlling the type I error rate.
Let’s illustrate how to use the Bonferroni adjustment for multiple comparisons implement in the
tgi_single_time function. This adjustment will ensure that we control the family-wise error
rate when comparing multiple treatment groups to the control.
In a typical linear regression model (with an intercept), the intercept represents the
average predicted value of the outcome variable when all predictor variables are zero. It serves as a
baseline reference point. For example, in the model log_tv ~ group, the intercept represents
the average log tumor volume for the reference group (typically the control group). The coefficients for
each treatment group then indicate how much their average log tumor volume differs from the control
group’s average.
When we add - 1 to the formula log_tv ~ group - 1, we’re explicitly telling the
model not to include an intercept term. This has two key effects:
Change in interpretation: the coefficients now represent the absolute average log tumor volumes
for each group, not the differences from a reference group. There’s no longer a baseline value that the
treatment effects are compared to.
Change in calculation: the model no longer tries to find a single intercept that best fits all groups.
Instead, it estimates separate intercepts for each group, essentially allowing each group’s average log
tumor volume to vary independently.
Imagine we have two groups with the following log tumor volumes:
Control: [4.5, 4.6, 4.7]
Treatment: [4.2, 4.3, 4.4]
The intercept \(\alpha\) (which is different from the \(\alpha_1, \alpha_2, \dots, \alpha_J\) coefficients defining the TGI model)
might be estimated as 4.6 (the average of the control group), and the coefficient for the treatment group
\(\beta\) would be -0.3 (the average difference from the control group),
using a model with intercept. With a model without intercept, the coefficients would be estimated directly
as the mean of each group, i.e., 4.6 for the control group, and 4.3 for the treatment group.
If there’s a strong theoretical reason to believe that the outcome variable should be zero when all
predictors are zero, we might want to force a zero intercept.
Code
# Example dataset for this demonstrationtv_data = TGI_dataset[ (TGI_dataset['group'].isin(['therapy_b', 'control']))].copy()# Log transformationtv_data['log_tv'] = np.log(tv_data['tv'])# Putting the baseline tumor volume in columntv_data['log_tv_baseline'] = ( tv_data .groupby('animal') # we pick for each animal .log_tv # the log_tv value .transform('first') # of the first day, i.e., day 0)# Keeping only the data point we want to analyzetv_data = tv_data[tv_data['day'] ==18]tv_data.head()
day
animal
group
tv
log_tv
log_tv_baseline
6
18
control_1
control
2447.881533
7.802978
4.652302
13
18
control_2
control
1586.124262
7.369049
4.577525
20
18
control_3
control
2681.138968
7.893997
4.612758
27
18
control_4
control
463.809975
6.139475
4.634028
34
18
control_5
control
997.884643
6.905638
4.759049
Code
# First model that reproduces the default behavior of `tgi_single_time` (adjusted)smf.ols( formula="log_tv ~ log_tv_baseline + C(group, Treatment(reference='control'))", data=tv_data).fit().summary2().tables[1]
# Second model with intercept removedsmf.ols( formula="log_tv ~ log_tv_baseline + group - 1", data=tv_data).fit().summary2().tables[1]
Coef.
Std.Err.
t
P>|t|
[0.025
0.975]
group[control]
4.586897
3.325829
1.379174
0.179159
-2.237141
11.410934
group[therapy_b]
3.205567
3.305763
0.969691
0.340808
-3.577298
9.988432
log_tv_baseline
0.721260
0.718649
1.003633
0.324466
-0.753286
2.195806
4.1.7 Pre-calculating individual TGI-delta and running OLS without
adjustment
In this approach, we would calculate the difference between the tumor volume at the time point of interest
and the baseline tumor volume (and then log-transform these differences), for each animal, and then fit a
simple linear model using only the TGI-delta values as the outcome and the ‘group’ as the predictor. In
fact, creating a new column with adjusted values or ΔTGI is often preferred, as it can make the code more
readable and easier to interpret. Instead of adding ‘log_tv_baseline’ to the model formula, we create a new
column ‘log_tv_adjusted’ within the DataFrame itself. This column holds the log-transformed tumor volumes
with the baseline values subtracted for each animal. The model formula is simplified to
log_tv_adjusted ~ group when baseline adjustment is applied.
With baseline adjustment, \(\text{log\_tv} = \alpha + \beta_1 *
\text{log\_tv\_baseline} + \beta_2 * \text{group} + \epsilon\), where \(\beta_1\) represents the association between the baseline log tumor volume
and the final log tumor volume, \(\beta_2\) represents the average
difference in log tumor volume between the treatment and control groups, after accounting for the baseline
differences. The model will fit a plane through the data points, estimating \(\alpha\), \(\beta_1\), and \(\beta_2\). In this case, \(\beta_2\)
isolates the effect of the treatment, controlling for the baseline.
Mathematically, the model with ‘log_tv_baseline’ effectively adjusts for the baseline within the regression
framework itself. By including ‘log_tv_baseline’ as a covariate, the model is implicitly calculating and
adjusting for the differences in baseline tumor volumes. The individual TGI-delta values that we would
calculate in the second approach are essentially derived from the same underlying relationship that
the first model captures. The TGI-delta is simply the difference between the log tumor volume at a specific
time point and the baseline log tumor volume, which is exactly what the ‘log_tv_baseline’ term is accounting
for.
The second approach (pre-calculating TGI-delta) might be slightly easier to understand conceptually, as the
adjustment is more explicit. However, the first approach (including ‘log_tv_baseline’ in the model) is often
more statistically sound and offers more flexibility for modeling complex relationships. Moreover, the
relationship between baseline tumor volume and the outcome is linear on the log scale, then both approaches
should give similar results in terms of statistical power.
Code
# Precalculated individual TGI-delta# Example dataset for this demonstration (fresh copy)tv_data = TGI_dataset[ (TGI_dataset['group'].isin(['therapy_b', 'control']))].copy()# Putting the baseline tumor volume in columntv_data['tv_baseline'] = ( tv_data .groupby('animal') # we pick for each animal .tv # the raw tv value .transform('first') # of the first day, i.e., day 0)tv_data['tv_adjusted'] = tv_data['tv'] - tv_data['tv_baseline']# Keeping only the data point we want to analyzetv_data = tv_data[tv_data['day'] ==18]# Pseudocount to avoid division by zeroespilon =1e-8# Log transformationtv_data['log_tv_adjusted'] = np.log(tv_data['tv_adjusted']+espilon)smf.ols(formula="log_tv_adjusted ~ group", data=tv_data).fit().summary2().tables[1]
Coef.
Std.Err.
t
P>|t|
[0.025
0.975]
Intercept
7.863679
0.217587
36.140394
4.855426e-25
7.417972
8.309385
group[T.therapy_b]
-1.555499
0.307714
-5.055007
2.386084e-05
-2.185823
-0.925174
4.1.8 Going beyond simple comparisons: ANCOVA and non-parametric
methods
In real-world preclinical studies, tumor growth can be influenced by more than just the treatment itself.
Factors like the initial tumor size, drug dosage, or even animal characteristics (sex, weight) can play a
role. To account for these additional variables (covariates), we can extend our simple
linear regression model to an analysis of covariance (ANCOVA) model.
4.1.8.1 Analysis of covariance (ANCOVA)
Purpose: adjusts the estimated treatment effects for the influence of covariates.
Model: the ANCOVA model includes both the treatment group and the relevant covariates as predictors:
\[{y}_i^{\ast
}={\sum}_{s=1}^S{\beta}_s{x}_{is}+{\sum}_{j=1}^J{\alpha}_j{I}_{ij}+{\epsilon}_i\] where \({\epsilon}_i\sim N\left(0,{\sigma}^2\right)\), \({x}_{si}\) represents the \(i\)
observations for the covariate variable \({\boldsymbol{X}}_S\) (e.g.,
log-transformed tumor volumes measured at baseline, dose levels of a treatment, or animal sex) and \({\beta}_s\) models the effects of the covariate on the final tumor
volume.
Benefits: improves the accuracy of treatment effect estimates by reducing unexplained variability and
increasing statistical power. We can then use the estimated parameters \(\hat{\alpha_1}\) (the intercept) and \(\hat{\alpha_T}\) (the treatment effect) in the original TGI-delta
formula, now using the adjusted treatment effect from the ANCOVA model. We obtain a more accurate and
statistically sound estimate of the TGI-delta, helping us isolate the true effect of the treatment on
tumor growth while accounting for any initial differences in tumor size among the experimental groups.
Example: if baseline tumor volume is a covariate, ANCOVA can more accurately estimate how much the
treatment affects tumor growth beyond the initial size differences.
Implementation: in Python, we can add covariates to the statsmodels model formula, as we
did in the previous examples using ‘log_tv_baseline’.
4.1.8.2 Handling unequal variances
In the standard linear regression model (and ANOVA), we assume that the variance of the errors (the
spread of the data around the fitted line) is constant across all groups. This is known as
homoscedasticity. However, in some TGI studies, the variability of tumor volumes might differ between
treatment groups. This is called heteroscedasticity. Ignoring heteroscedasticity can lead
to inaccurate estimates of standard errors, confidence intervals, and P-values, potentially leading to
incorrect conclusions about treatment effects.
To address this issue, we can relax the assumption of equal variances by allowing each group to have its
own variance: \({\epsilon}_i\sim N\left(0,\boldsymbol{\Sigma} \right)\).
The matrix representation of the residual error variance \(\boldsymbol{\Sigma}\) provides a way to do this:
In this matrix, the diagonal elements (\(\sigma_1^2, \sigma_2^2, \dots,
\sigma_J^2\)) represent the variances of the residual errors within each of the \(J\) treatment groups, and the off-diagonal elements are all zero,
indicating that the errors between different groups are assumed to be uncorrelated.
This matrix form allows each group to have its own unique variance, capturing the potential
heteroscedasticity in the data. It is used in a technique called weighted
least squares (WLS) regression. In WLS, each observation is weighted by the inverse of its estimated
variance. This gives more weight to observations with lower variance (more precise measurements) and less
weight to observations with higher variance (less precise measurements). By properly accounting for the
unequal variances, WLS regression can provide more accurate estimates of the treatment effects and their
standard errors, leading to more reliable statistical inferences.
Code
# Keeping only the data point we want to analyzetv_data_all = TGI_dataset[TGI_dataset['day'] ==18].copy()# Log transformationtv_data_all['log_tv'] = np.log(tv_data_all['tv'])# Create a weight matrix# Add a small value to avoid zero variancesepsilon =1e-8# A small constant to prevent division by zero# We might want to use the population variance (ddof = 0) if we consider# each group to represent its own populationweights =1.0/ (tv_data_all.groupby('group') ['log_tv'].transform('var', ddof=0) + epsilon)# Fit weighted least squares modelsmf.wls( formula="log_tv ~ group", data=tv_data_all, weights=weights).fit().summary2().tables[1]
Coef.
Std.Err.
t
P>|t|
[0.025
0.975]
Intercept
7.919420
0.195191
40.572714
3.267968e-43
7.528406
8.310434
group[T.therapy_a]
-0.883228
0.233706
-3.779235
3.835349e-04
-1.351396
-0.415060
group[T.therapy_b]
-1.401502
0.267171
-5.245708
2.468133e-06
-1.936710
-0.866294
group[T.therapy_combo_ab]
-2.502872
0.276547
-9.050442
1.494836e-12
-3.056862
-1.948882
4.1.8.3 Non-parametric methods
When the assumptions of normality or equal variance are not met, or when there are significant outliers,
non-parametric tests provide a robust alternative for comparing tumor growth inhibition (TGI) between
treatment groups. Non-parametric tests don’t rely on specific assumptions about the
underlying distribution of the data. They compare the ranks of the tumor volumes across groups, rather
than the actual values.
The Wilcoxon-Mann-Whitney test is used when comparing two groups (\(J=2\)), e.g., treatment vs. control, and the Kruskal-Wallis test is
used when comparing more than two groups (\(J \gt 2\)).
Non-parametric tests are less sensitive to outliers and deviations from normality compared to parametric
tests, they can be more efficient than parametric tests when the normality assumption is violated and they
can be used with small sample sizes where normality tests might not be reliable. However, they are
generally less flexible for adjusting for covariates (e.g., baseline tumor volume), they are primarily
useful for testing hypotheses about differences between groups, not for estimating effect sizes or making
predictions, and while they don’t assume normality, they do require some assumptions, such as similar
distribution shapes across groups and equal variances.
4.2 Analyzing continuous data longitudinally
In contrast to single time-point studies, longitudinal TGI studies collect tumor volume measurements at
multiple points throughout the experiment. This allows researchers to analyze how tumor growth changes over
time.
Longitudinal data introduces a time component \(t\), where each tumor volume
measurement \(y_{ijt}\) is associated with a specific animal \(i\), treatment group \(j\), and time point
\(t\), where \(t = 1, \dots, D\) and \(D\) indicates the last time-point of data collection. The statistical analysis
approach depends on whether the time-dependent nature of the data is considered important or not.
Time-independent analysis: if the focus is solely on comparing treatment groups at each individual time
point (not the overall growth trend), we can analyze each time point separately using the same
methods as in single time-point studies (e.g., linear models, t-tests, ANOVA). This approach ignores the
potential dependency between measurements taken on the same animal over time.
Time-dependent analysis: if we want to understand the overall patterns of tumor growth over time and how
treatments affect those patterns, we’ll need to use more advanced statistical techniques that explicitly
model the time component.
If the variability in tumor growth within each group is very large, it might be difficult to
model the time-dependent trends accurately. In such cases, analyzing each time point separately might be a
simpler and more practical approach. If we’re primarily interested in comparing treatment effects at specific
key time points (e.g., end of study, midway through), we might not need to model the entire growth curve.
When baseline tumor volume is included as a covariate in the analysis (ANCOVA), the focus shifts to comparing
the tumor growth trajectories from the second time point onwards, with \(t = 2,
\dots, D\) where \(2\) is the second time-point of data collection.
4.2.1 Hypothesis testing
In treatment vs. control testing, for each time point, one-sided tests are typically used to assess
if a treatment reduces tumor volume compared to the control. (H1: \(\alpha_{jt}
\lt \alpha_{1t}\))
In pairwise treatment comparisons, two-sided tests are used to compare the efficacy of different
treatments at each time point. (e.g., H1: \(\alpha_{2t} \ne
\alpha_{3t}\))
Since multiple hypotheses are being tested (one for each time point and treatment comparison), it’s crucial
to adjust for multiple comparisons to control the overall Type I error rate (false positives).
Bonferroni correction: a simple but conservative approach that divides the significance level by the
number of comparisons.
Tukey’s honestly significant difference (HSD): a more powerful method often used when sample sizes are
balanced across groups.
Duncan’s multiple range test: another option for pairwise comparisons.
Dunnett’s test: when all treatments are compared against a common reference (control), Dunnett’s test is
a specialized method that accounts for the correlation between the comparisons and is generally more
powerful than other adjustments.
4.2.2 TGI analysis over multiple time points
Unlike single-time-point analysis, assessing tumor growth inhibition (TGI) over multiple time points
provides a more comprehensive view of treatment efficacy. This longitudinal approach allows us to track how
tumor volumes change in response to treatment over the entire study duration, offering insights into the
dynamics of tumor growth and the potential for long-term therapeutic effects.
To analyze TGI across multiple time points, we’ll adapt our existing tgi_single_time function.
This modified function will iterate through each specified time point, fitting a linear model at each and
calculating the corresponding TGI.
Code
from scipy.stats import dunnettdef tgi_multiple_time( dataset: pd.DataFrame, time_points: List[int], treatments_numerator: List[str], treatment_denominator: str='control', baseline_adjusted: bool=False, baseline: int=0, p_tails: int=1, correction_method: str='none',) -> pd.DataFrame:""" Estimates Tumor Growth Inhibition (TGI) at multiple time points using linear models. Args: dataset (pd.DataFrame): DataFrame with columns 'day', 'group', and 'tv' (tumor volume). time_points (List[int]): List of days at which to calculate TGI. Must have at least 2 time points. treatments_numerator (List[str]): List of treatment group names. treatment_denominator (str, optional): Name of the comparison group. Defaults to 'control'. baseline_adjusted (bool, optional): If True, adjust for baseline tumor volume. Defaults to False. baseline (int, optional): Baseline day for adjustment. Defaults to 0. p_tails (int, optional): Number of tails for hypothesis test (1 for one-sided, 2 for two-sided). Defaults to 1. correction_method (str, optional): Method for multiple comparisons correction ('none', 'bonferroni', 'sidak', 'dunnett'). Defaults to 'none'. Returns: pd.DataFrame: DataFrame containing TGI estimates, confidence intervals, and P-values for each treatment at each time point. """iflen(time_points) <2:raiseValueError("Please provide at least 2 time points for TGI calculation across multiple time points.")# Filter data for relevant groups and time points (including baseline if adjusting) relevant_days = time_points +\ [baseline] if baseline_adjusted else time_points tv_data = dataset[ (dataset['day'].isin(relevant_days))& (dataset['group'].isin(treatments_numerator + [treatment_denominator])) ].copy()# Log-transform tumor volume tv_data['log_tv'] = np.log(tv_data['tv'])# Baseline adjustmentif baseline_adjusted: tv_data['log_tv_baseline'] = tv_data.groupby('animal')['log_tv'].transform('first')# Convert Day to categorical, not simply integers, with correct order tv_data['day'] = pd.Categorical( tv_data['day'], categories=sorted(time_points), ordered=True)# Results container model_estimates_list = []# Iterate over time pointsfor time_point in time_points:# Filter data for the current time point data_time_point = tv_data[tv_data['day'] == time_point]# Fit the linear modelif baseline_adjusted: model = smf.ols( formula=f"log_tv ~ log_tv_baseline + C(group, Treatment(reference='{treatment_denominator}'))", data=data_time_point)else: model = smf.ols( formula=f"log_tv ~ C(group, Treatment(reference='{treatment_denominator}'))", data=data_time_point) est = model.fit()# Extract model estimates and calculate TGI for each treatment group results = est.summary2().tables[1]# print(results)for treatment in treatments_numerator:# Extract TGI, CI, and P value for the current treatment ix =f"C(group, Treatment(reference='{ treatment_denominator}'))[T.{treatment}]" tgi = (1- np.exp(results.loc[ix, "Coef."]))# Calculate confidence intervals and P value std_err = results.loc[ix, "Std.Err."] degrees_of_freedom = model.df_residif p_tails ==1: lb95_tgi = np.nan ub95_tgi = (1- np.exp(results.loc[ix, "Coef."] + t_dist.ppf(0.95, degrees_of_freedom) * std_err)) pvalue = t_dist.cdf(results.loc[ix, "t"], degrees_of_freedom)else: lb95_tgi = (1- np.exp(results.loc[ix, "Coef."] + t_dist.ppf(0.025, degrees_of_freedom) * std_err)) ub95_tgi = (1- np.exp(results.loc[ix, "Coef."] + t_dist.ppf(0.975, degrees_of_freedom) * std_err)) pvalue =2*\ (1- t_dist.cdf(abs(results.loc[ix, "t"]), degrees_of_freedom))# Adjust p-values if multiple comparison correction is specifiedif correction_method =='bonferroni': pvalue *=len(treatments_numerator)elif correction_method =='sidak': pvalue =1- (1- pvalue) **len(treatments_numerator)# Create a dataframe of the results for the current treatment model_estimates = pd.DataFrame({"contrast": [f"{treatment} vs. {treatment_denominator}"],"day": [time_point],"tgi": [tgi],"lb95_tgi": [lb95_tgi],"ub95_tgi": [ub95_tgi],"pvalue": [pvalue],"df": [degrees_of_freedom],"baseline_adj": [baseline_adjusted],"correction": [correction_method], })# Append results for this treatment to the list model_estimates_list.append(model_estimates)# Combine results into a single DataFrame all_results = pd.concat(model_estimates_list, ignore_index=True)# scipy.stats >= 1.11 includes Dunnett's test for multiple comparisons# Apply Dunnett's test# TO DO: DOUBLE CHECK THE RESULTS OF THE DUNNETT'S CORRECTION# TO DO: IMPLEMENT DUNNETT'S CORRECTION IN `tgi_single_time`?if correction_method =='dunnett': control_data = tv_data[tv_data['group'] == treatment_denominator]['log_tv'].values treatment_data = [tv_data[tv_data['group'] == trt] ['log_tv'].values for trt in treatments_numerator] dunnett_result = dunnett(*treatment_data, control=control_data)# Update P-values in the results DataFramefor i, trt inenumerate(treatments_numerator): all_results.loc[all_results["contrast"] ==f"{trt} vs. { treatment_denominator}", "pvalue"] = dunnett_result.pvalue[i]# Combine results into a single DataFramereturn all_results
To gain a deeper understanding of how our various treatments affect tumor growth throughout the study,
we’ll perform a multiple time point analysis using the tgi_multiple_time function. This will
allow us to track changes in tumor growth inhibition (TGI) over the entire duration of the experiment and
compare the effectiveness of different therapies at each measurement time point.
We specify all the time points at which we want to analyze TGI, including day 0 (baseline). In this
example, we’re using the default values for ‘baseline_adjusted’ (False), ‘p_tails’ (1 for one-sided tests),
and ‘correction_method’ (no correction).
This analysis provides a more comprehensive view of treatment efficacy compared to single-time-point
analysis. By analyzing multiple time points, we can identify potential trends and changes in treatment
effects over the study duration. This example doesn’t include baseline adjustment or multiple comparisons
correction. These can be added as needed based on the specific research question and study design, as in the
following example.
The Bonferroni adjustment divides the desired significance level (e.g., 0.05) by the number of comparisons.
In this case, we have 6 time points and 3 treatment groups, so 18 comparisons in total. This means the
adjusted significance level for each individual test would be approximately 0.0028.
The P values obtained after Bonferroni correction are generally higher compared to Dunnett’s correction.
When using Bonferroni, we’ll typically find fewer statistically significant results compared to using
Dunnett’s test. This is because Bonferroni is more stringent and requires stronger evidence to reject the
null hypothesis. If we are specifically interested in comparing multiple treatment groups to a single
control, and we are confident that the variances are roughly equal across groups, then Dunnett’s test is
often a better choice as it provides more statistical power while still controlling the family-wise error
rate (FWER).
Now, let’s focus on a direct comparison between two treatment groups: ‘therapy_a’ and ‘therapy_b’. It
doesn’t involve the control group. This type of analysis is often done to assess whether a new treatment is
non-inferior or superior to an existing standard therapy. The function calculates TGI (adjusted for
baseline) at multiple time points (days 3, 6, 9, 12, 15, and 18). This allows us to track how the difference
in efficacy between both treatments evolves over the course of the study.
The inclusion of baseline_adjusted=True (with baseline=0) indicates that the
analysis will account for potential differences in the initial tumor volumes between the two groups at day
0. This is crucial when comparing two active treatments, as baseline differences could confound the results.
By setting p_tails=2, we are conducting two-sided hypothesis tests at each time point. This
means we’re looking for evidence that ‘therapy_a’ is either significantly better or significantly worse than
‘therapy_b’. This is different from the usual one-sided tests used when comparing a treatment to a control,
where the focus is on whether the treatment is better. In this specific example, we’re not applying any
correction for multiple comparisons (e.g., Bonferroni or Dunnett’s). This is because we are primarily
interested in the time course of the comparison between the two treatments, not just the individual P-values
at each time point. However, we might consider adding a correction if we want to make more definitive
conclusions about the overall significance of the differences across all time points.
4.2.3 Why the degrees of freedom don’t increase as expected
The reason why the degrees of freedom don’t increase as we would expect in a true repeated measures
analysis is that we are not explicitly modeling the correlation structure of the repeated measurements over
time within each animal. Instead, we are treating each time point as an independent observation, leading to
separate analyses for each time point.
This approach has some important implications:
Inflated degrees of freedom: the calculated degrees of freedom might be overestimated because the
repeated measures within animals are not accounted for. This could lead to overly optimistic P-values and
potentially false-positive findings.
Reduced power: by not considering the correlation structure, we may be missing out on potential gains in
statistical power that could come from utilizing the information contained in the repeated measures.
Limited inferences: the results of this analysis are specific to each individual time point and might
not provide a comprehensive picture of the overall treatment effect over time.
To address these issues, we have a few options:
Repeated measures ANOVA: this is a more appropriate statistical method for analyzing longitudinal data,
as it explicitly models the correlation structure of repeated measures. However, it requires certain
assumptions about the correlation structure and might not be suitable for all types of data.
Mixed effects models: these models offer greater flexibility in modeling complex correlation structures
and are often a good choice for analyzing longitudinal data. They allow us to account for both fixed
effects (treatment groups) and random effects (individual animal variations).
Generalized estimating equations (GEE): GEE models provide a robust way to analyze longitudinal data
when the correlation structure is complex or unknown. They can be more flexible than repeated measures
ANOVA and are less sensitive to missing data.
4.2.4 Analyzing longitudinal TGI data
4.2.4.1 Modeling tumor growth curves
When researchers are interested in understanding the dynamics of tumor growth over time, longitudinal TGI
studies provide a valuable framework. By summarizing tumor growth curves into a few key parameters, such
as growth rate or area under the curve, we can make meaningful comparisons between treatment groups.
If the variability between individual tumors within a group isn’t too large, we can use longitudinal
models to analyze the data. This approach is generally more efficient than looking at single time points
because it takes into account the entire growth pattern over time.
For short-term TGI studies (common due to animal welfare considerations), the exponential
model is often used to fit the growth curves. This model assumes that tumors grow
exponentially, translating to a linear relationship between log-transformed tumor volumes and time. Mixed-effects models (MEMs) are a
powerful statistical framework for analyzing longitudinal data. In the context of TGI studies, they allow
us to estimate the average growth curve for each treatment group while accounting for individual
variations in tumor growth between animals:
Fixed effects: represent the average intercept and slope (growth rate) for each treatment
group.
Random effects: represent the individual deviations in intercept and slope for each animal within a
treatment group.
Error structure: models both between-animal (variations between animals in the same group) and
within-animal (variations in measurements for the same animal) sources of variability.
By accounting for within-animal variability (correlations between measurements from the same animal),
mixed-effects, i.e., fixed + random, models are more efficient than fixed-effects models, which ignore
this correlation. Furthermore, the model’s inferences can be extended to the broader population of
animals, not just the specific sample in the study.
\(\stackrel{*}{y_{it}}\): log-transformed tumor volume for animal
\(i\) at time \(t\).
\(t_{ij}\): time of measurement for animal \(i\) in group \(j\).
\(\alpha_j\): intercept (initial tumor volume) for treatment group
\(j\).
\(\omega_{ij}\): random intercept deviation for animal \(i\) in group \(j\).
\(\beta_j\): slope (growth rate) for treatment group \(j\).
\(\varphi_{ij}\): random slope deviation for animal \(i\) in group \(j\).
\(I_{ij}\): indicator variable (\(1\) if animal \(i\) is in group \(j\), \(0\) otherwise).
\({\Sigma}_{it}\): residual error term.
4.2.4.2.1 Random effects: capturing individual animal variations
The random intercept (\(\omega\)) represe_nts the baseline
differences in tumor volume between individual animals within the same treatment group.
Some animals might naturally have larger or smaller tumors at the start, regardless of the treatment.
The random slope (\(\varphi\)) represents the individual
differences in tumor growth rates within the same treatment group. Some animals might
naturally have faster or slower growing tumors than others in their group.
The variance-covariance matrix \(\boldsymbol{G}\)
describes the relationships between these random effects \(\boldsymbol{G}=\left[\begin{array}{cc}{\sigma}_{\omega}^2& {\sigma}_{\omega
\varphi}^2\\ {}{\sigma}_{\omega \varphi}^2& {\sigma}_{\varphi}^2\end{array}\right]\), where
\({\sigma}_{\omega}^2\) is the variance of the random intercepts
(between-animal variability in initial tumor volume), \({\sigma}_{\varphi}^2\) is the variance of the random slopes
(between-animal variability in growth rate), and \({\sigma}_{\omega
\varphi}^2\) is the covariance between random intercepts and slopes. This captures whether
animals with larger initial tumors tend to have faster or slower growth rates.
4.2.4.2.2 Between-animal vs. within-animal variation
The between-animal variation is the variation in tumor growth between different
animals within the same treatment group. It’s captured by the random effects and their
variance-covariance matrix (\(\boldsymbol{G}\)). The
within-animal variation is the variation in tumor growth within the same animal over
time. It’s captured by the residual error term (\({\Sigma}_{it}\)). In
the context of the mixed effects model for tumor growth, the residual error term captures the
variability in tumor volume that is not explained by the fixed effects (treatment group effects) or the
random effects (individual animal deviations). Specifically, \({\Sigma}_{it}\) might be modeled as a combination of:
Measurement error: the inherent variability in measuring tumor volume, which could be due to the
limitations of the measurement technique or biological factors.
Within-animal variability: the natural fluctuations in tumor growth over time within an individual
animal that are not captured by the overall growth trend.
4.2.4.2.3 Advantages of MEMs
Mixed effects models are particularly well-suited for analyzing longitudinal TGI data because they:
Account for correlation: they explicitly model the correlation between repeated measurements on the
same animal, recognizing that these measurements are not independent.
Increased efficiency: by accounting for within-animal variability, they can estimate the treatment
effects more efficiently, potentially leading to higher statistical power.
Population-level inference: unlike fixed effects models, mixed effects models allow us to make
inferences about the larger population of animals, not just the specific individuals in the study.
Handling missing data: they can handle missing data points more robustly than traditional repeated
measures ANOVA, as long as the missingness is random.
4.2.4.3 Estimating TGR with MEMs with Python
4.2.4.3.1 Statistical distributions
R’s nlme::lme function uses t-tests to assess the significance of the fixed effects,
particularly when the sample size is relatively small or when the degrees of freedom for the denominator
of the test statistic are not well-defined. The t-distribution is appropriate in these cases because it
accounts for the uncertainty in estimating the standard error of the fixed effects. R often defaults to
reporting t-values in model summaries, even if the sample size is large enough that a z-test would be
appropriate. This is likely due to historical reasons and the common practice of using t-tests for small
samples.
Statsmodels’ mixedlm, on the other hand, tends to use z-tests (based on the
standard normal distribution) to assess the significance of the fixed effects. This is often a
reasonable approximation when the sample size is large, and the degrees of freedom for the
denominator are well-estimated. Statsmodels generally favors reporting z-values in model summaries, as
it assumes that you are often working with larger sample sizes where the z-test is a suitable
approximation.
While the test statistics (t-values vs. z-values) might differ slightly, the interpretation of the
results remains similar. The P-values associated with these test statistics can be used to assess the
statistical significance of the fixed effects in both cases. If the sample size is small, the
t-distribution might be a more appropriate choice. We could consider calculating the degrees
of freedom and use t-distribution table (or Python’s scipy.stats module) to obtain more accurate
p-values. If the sample size is large, the
difference between t-values and z-values will be negligible. In most practical cases, the
difference in results is negligible.
In the mixed-effects model used for TGR analysis, the degrees of freedom are calculated based on the
number of animals (groups) and the number of time points. Since we typically have multiple time points,
this often results in a relatively high number of residual degrees of freedom. For example, in a study
with one treatment group and one control group with 6 animals per group, and 6 time-points analyzed, the
degrees of freedom exceed 30.
If the number of animals and/or time points is very small, or in some complex mixed-effects models
where the exact degrees of freedom might not be easily determined, using the t-distribution can be a
more conservative approach in such cases.
4.2.4.3.2 The interaction effect
In the model, the formula log_tv ~ C(group, Treatment(reference='control')) * day
specifies a model with C(group, Treatment(reference='control')) which represents the effect
of each treatment group (excluding the reference group, ‘control’) on the log tumor volume, i.e., the
main effects, and * day the interaction term which models how the effect
of each treatment group changes over time, i.e., the interaction effect.
The main effect coefficient for C(group, Treatment(reference='control'))[T.therapy_xx]
only captures the average difference in tumor growth rate between the ‘therapy_xx’ group and
the control group across all time points. However, the treatment effect might not be constant over time;
it could increase or decrease as the study progresses.
In a TGR analysis, we’re interested in the rate of change of tumor volume over time, not just
the overall difference at a specific time point. The interaction term group:day captures
this rate of change (slope) for each treatment group relative to the control group.
Therefore, by extracting the coefficient from
C(group, Treatment(reference='control'))[T.therapy_xx]:day, we’re getting the estimated
difference in the tumor growth rate between the ‘therapy_xx’ group and the ‘control’ group, i.e.,
the TGR value. Let’s say the coefficient for the interaction term is -0.05. This means that, on average,
for each additional day in the study, the ‘therapy_xx’ group’s log tumor volume is estimated to be 0.05
units lower than what would be expected based on the control group’s growth rate and the overall
treatment effect of “therapy_xx.”, in other words, the therapy’s effectiveness in reducing tumor growth
increases over time compared to the control group.
Code
import warningsfrom statsmodels.tools.sm_exceptions import ConvergenceWarningwarnings.simplefilter('ignore', ConvergenceWarning)# These are warnings that we show as a way of alerting you that we may be in a non-standard situation. Most likely, one of# the variance parameters is converging to zero. If we get a model result and the parameter estimates and standard errors are# finite, we can just use them and ignore all the warnings. Note that inference for variance parameters is not meaningful when# the point estimate is at or near zero.def tumor_growth_curves_exponential( dataset: pd.DataFrame, treatments_numerator: List[str], treatment_denominator: str='control', time_points: List[int] =None, p_tails: int=1) -> pd.DataFrame:""" Estimates and compares tumor growth rates (TGR) between treatment groups using a linear mixed-effects model. Args: dataset (pd.DataFrame): DataFrame with columns 'day', 'group', and 'tv' (tumor volume). treatments_numerator (List[str]): List of treatment group names. treatment_denominator (str, optional): Name of the comparison group. Defaults to 'control'. time_points (List[int], optional): List of days at which to calculate TGR. If None, uses all unique days. p_tails (int, optional): 1 for one-sided test (tumor reduction vs. control), 2 for two-sided test. Defaults to 1. Returns: pd.DataFrame: DataFrame containing TGR estimates, confidence intervals, and P-values for each contrast. """# Filter the dataset for relevant groups and time points included_groups = treatments_numerator + [treatment_denominator]ifnot time_points:# uses all unique days in the dataset time_points = dataset['day'].unique() tv_data = dataset[ (dataset['group'].isin(included_groups))& (dataset['day'].isin(time_points)) ].copy()# Log-transform tumor volume tv_data['log_tv'] = np.log(tv_data['tv'])# Convert 'day' to numeric and 'group' to categorical variable (good practice) tv_data['day'] = pd.to_numeric(tv_data['day']) tv_data['group'] = pd.Categorical( tv_data['group'], categories=[treatment_denominator] + treatments_numerator)# Fit mixed effects model (similar to lme in R) model = smf.mixedlm(# equivalent to R formula 'lnTV ~ Group * Day, random = ~ 1 + Day | Animal'# log_tv modeled as a function of group, day, and their interactionf"log_tv ~ C(group, Treatment(reference='{ treatment_denominator}'))*day", data=tv_data,# random effects# 'animal' is the grouping variable for the random effects groups=tv_data['animal'], re_formula='~day', # including a random slope for day for each animal# the mixedlm function automatically includes a random intercept term ) est = model.fit( reml=True, # default method=["lbfgs"] )# Extract model estimates for the interaction terms (group:day)# print(est.summary()) results = est.summary().tables[1]# Need to trick a little bit the outputfor col in results.columns: results[col] = pd.to_numeric(results[col], errors='coerce')# print(results)# Results container model_estimates_list = []for treatment in treatments_numerator:# Extract TGI, CI, and P value for the interaction terms (group:day) ix =f"C(group, Treatment(reference='{ treatment_denominator}'))[T.{treatment}]:day" tgr = results.loc[ix, "Coef."]# Calculate confidence intervals and P value std_err = results.loc[ix, "Std.Err."]if p_tails ==1: lb95_tgr = np.nan ub95_tgr = results.loc[ix, "Coef."] + norm.ppf(0.95) * std_err# Although the t-distribution is used in R pvalue = norm.cdf(results.loc[ix, "z"])# we just pickup the results from statsmodels as it (normal distribution)else: lb95_tgr = results.loc[ix, "[0.025"] ub95_tgr = results.loc[ix, "0.975]"] pvalue = results.loc[ix, "P>|z|"]# Create a dataframe of the results for the current treatment model_estimates = pd.DataFrame({"contrast": [f"{treatment} vs. {treatment_denominator}"],"time_window": [f"{tv_data['day'].min()}-{tv_data['day'].max()}"],"tgr": [tgr],"lb95_tgr": [lb95_tgr],"ub95_tgr": [ub95_tgr],"pvalue": [pvalue], })# Append results for this treatment to the list model_estimates_list.append(model_estimates)# Combine results into a single DataFramereturn pd.concat(model_estimates_list, ignore_index=True)
4.2.4.3.3 Examples
In the first example, the analysis aims to assess whether the tumor growth rates (TGR) of three
different therapies (A, B, and their combination) are significantly lower than the control group’s TGR
across multiple time points. We’re interested in tumor reduction (negative TGRs) compared to the
control, so a one-sided test is appropriate. And we’re analyzing the entire time window of the study
(days 0 to 18) to capture the full dynamics of tumor growth.
The TGR values represent the estimated average difference in the growth rates of each
treatment group compared to the control group. For example, a negative TGR for “therapy_a
vs. control” means that ‘therapy_a’ is associated with slower tumor growth than the control. The P
value tells us the probability of observing a TGR as extreme as (or more extreme than) the one we
obtained, assuming the null hypothesis (no difference in growth rates) is true. A smaller P value (e.g.,
less than 0.05) suggests that the observed difference is statistically significant and unlikely due to
chance. Since this is a one-sided test, we would only focus on negative values of TGR that are
significant.
This analysis allows us to see which treatments are significantly more effective than the control group
in slowing down tumor growth over the entire study duration. We can also compare the
relative efficacy of different treatments by looking at their TGR values and confidence intervals.
In the second example, we compare ‘therapy_a’ to ‘therapy_a’, focusing on a specific time window (days
9-18). The exponential growth model assuming that the log-transformed tumor volumes increase linearly
with time, if this assumption is violated (e.g., due to non-linear growth patterns), the model might not
be appropriate, and the results could be misleading. By selecting time points where the linearity
assumption holds, we improve the validity of the analysis.
This analysis aims to answer the question: “is therapy_a non-inferior or superior to therapy_b in terms
of tumor growth rate over the selected time window (days 9-18)?”. We use a two-sided test because we
want to determine if therapy_a is either better or worse than therapy_b, not just if it’s better. And by
using only time points where the log tumor volumes exhibit relative linearity, we’re ensuring that the
exponential growth model is a good fit for the data and that the estimated TGRs are reliable.
The TGR estimate of 0.056 indicates that, on average, the tumor growth rate in the ‘therapy_a’ group is
5.6% higher per day than the tumor growth rate in the ‘therapy_b’ group over the analyzed time window.
This is the difference in the slopes of the log-transformed tumor volume curves between the two groups.
Let’s compare this result to the analysis of the entire time windows in the same groups.
The exponential growth model assumes that the logarithm of tumor volume increases linearly with time.
However, in reality, early tumor growth often doesn’t perfectly follow this pattern. There can be a
lag phase or initial slow growth before the exponential phase kicks in. Including these
early time points in the model can: -Bias the slope estimate: the early, slower growth can pull down the
estimated slope of the regression line, making it appear flatter than it actually is during the truly
exponential phase. This can underestimate the treatment effect (difference in slopes between groups).
Increase variability: the early time points might have more variability in tumor volume, which can
increase the residual error in the model and reduce statistical power.
Violate model assumptions: the exponential model assumes that the log-transformed tumor volume is
linearly related to time. If this assumption doesn’t hold in the early time points, the model fit will
be worse, potentially leading to inaccurate estimates and misleading conclusions.
By selecting time points where the log tumor volume appears to be linear (e.g., 6, 9, 12, 15, 18 days
in our example), we’re focusing the analysis on the period where the exponential growth assumption is
more likely to be valid. This can lead to more accurate TGR estimates, reduced variability, and better
model fit.
4.2.4.4 Modeling tumor growth with quadratic exponential models
In preclinical TGI studies that run for a moderate duration, a phenomenon known as tumor growth
deceleration might occur towards the end of the experiment. This means that the tumor growth
rate slows down, rather than remaining constant as assumed in the exponential growth model. This
deceleration can be attributed to various factors:
Resource limitations: as tumors grow larger, they might outstrip the available resources in their
surrounding environment, such as nutrients and oxygen, leading to slower growth.
Spatial constraints: the physical space within the animal or the tumor implantation site might limit
the tumor’s ability to expand further.
Treatment effects: some treatments might not only inhibit tumor growth but also actively induce tumor
regression or cell death, causing a decline in the growth rate.
To account for this deceleration, a more flexible model is needed. The quadratic exponential model is a
suitable alternative as it incorporates a term that captures the change in growth rate over time. The
model is defined as:
\({Y}_{it}^{\ast}\): log-transformed tumor volume for animal \(i\) at time \(t\).
\({\alpha}_j\): intercept (initial tumor volume) for treatment group
\(j\).
\({\beta}_j\): initial growth rate for treatment group \(j\).
\({\gamma}_j\): tumor decay rate (TDR) for treatment group \(j\), capturing the deceleration effect over time.
\({t}_{ij}\): time of measurement for animal \(i\) in group \(j\).
\({I}_{ij}\): indicator variable (\(1\) if animal \(i\) is in group \(j\), \(0\) otherwise).
The quadratic term (\({t}_{ij}^2\)) allows for modeling a change in
growth rate over time. A positive \({\gamma}_j\) implies a decreasing
growth rate (deceleration), while a negative \({\gamma}_j\) would
indicate an increasing growth rate (acceleration). The longer the experiment, the more pronounced the
effect of the quadratic term becomes. This is because the square of the time (\({t}_{ij}^2\)) becomes larger, leading to a faster deceleration (or
acceleration) of tumor growth.
This model can better represent the complex dynamics of tumor growth, where the rate might not be
constant throughout the study. And by capturing the deceleration of growth, the model can lead to more
accurate estimates of treatment effects compared to a simple exponential model. However, the model is more
complex than the simple exponential model, requiring additional parameters (\({\gamma}_j\)) to be estimated, and the interpretation of the coefficients
might be less intuitive, as the growth rate is no longer constant but depends on time.
To simulate the complex, non-linear growth patterns observed in some tumors, the function below creates
virtual groups of animals whose tumor volumes evolve according to the quadratic exponential model, a more
nuanced approach than simple exponential growth.
Code
def simulate_quad_exp_growth( group_name: str, duration: int, initial_tumor_volume: float, group_growth_rate_day_pct: float, growth_rate_cv: float, group_decay_rate_day_pct: float, decay_rate_cv: float, residual_cv: float, n_animals: int, max_tumor_volume: float, df=None,) -> pd.DataFrame:""" Simulates tumor growth data with quadratic exponential growth for a group of animals. Args: group_name (str): Name of the group (e.g., "control", "therapy_a"). duration (int): Duration of the study in days. initial_tumor_volume (float): Initial tumor volume (mm^3). group_growth_rate_day_perc (float): Average daily growth rate of the group (%). growth_rate_cv (float): Coefficient of variation for the growth rate within the group. group_decay_rate_day_perc (float): Average daily decay rate of the group (%). decay_rate_cv (float): Coefficient of variation for the decay rate within the group. residual_cv (float): Coefficient of variation for the measurement error. n_animals (int): Number of animals in the group. max_tumor_volume (float): Maximum allowed tumor volume before removal (mm^3). df (int, optional): Degrees of freedom for the t-distribution. If None, defaults to n_animals - 1. Returns: pd.DataFrame: Simulated data with columns: day, animal, group, tv (Tumor Volume). """# Initialize an empty list to store DataFrames for each animal in the group group_data = []# Loop over each animal in the groupfor i inrange(n_animals):# Create an array of time points (days) when measurements are taken, starting from# day 0 up to the end of the study (`duration`). Measurements are taken every 3 days. days = np.arange(0, duration +1, 3)# Sample individual growth and decay rates## growth_rate_individual = np.random.normal(# group_growth_rate_day_pct / 100,# group_growth_rate_day_pct * growth_rate_cv / 100,# len(days) - 1# )## Since we're simulating tumor growth data, and the sample size is usually small# (below 15 animals per group), using the t distribution could be a reasonable choice.# It will introduce slightly more variability into the simulated data, reflecting the# uncertainty associated with small samples and the stochastic nature of tumor growth.# Shift and scale the t-distribution for individual growth rate df = n_animals -1if df isNoneelse df # Default df if not provided growth_rate_individual = t_dist.rvs( df=df, loc=group_growth_rate_day_pct /100, scale=group_growth_rate_day_pct * growth_rate_cv /100, size=len(days) -1 )# Same for the average daily decay rate# decay_rate_individual = np.random.normal(# -group_decay_rate_day_pct / 100, # Note the negative sign for decay# group_decay_rate_day_pct * decay_rate_cv / 100,# len(days) - 1# ) decay_rate_individual = t_dist.rvs( df=df, loc=-group_decay_rate_day_pct /100, # Note the negative sign for decay scale=group_decay_rate_day_pct * decay_rate_cv /100, size=len(days) -1 )# Note: if the growth rates are strictly positive and tend to be right-skewed (some# tumors grow much faster than others), the gamma distribution can be a good option.# Initialize an empty array to store the tumor volumes over time ln_tumor_volume = np.empty(len(days))# ln.tumor.volume[1] <- rnorm(n = 1, mean = log(initial.tumor.volume), sd = sqrt(log((residual.cv^2) + 1))) ln_tumor_volume[0] = np.random.normal( loc=np.log(initial_tumor_volume), scale=np.sqrt(np.log(residual_cv**2+1)) )# Iterate over time points (days) to simulate tumor growthfor t inrange(1, len(days)): ln_tumor_volume[t] = np.random.normal( ln_tumor_volume[t -1] + growth_rate_individual[t -1] * days[t] + decay_rate_individual[t -1] * days[t]**2, np.sqrt(np.log(residual_cv**2+1)) )# Considere Gompertz Growth Model that describes sigmoidal growth patterns,# which are often observed in tumors.# # Transform log-tumor volumes back to the original scale (tumor volumes in mm^3). tumor_volume = np.exp(ln_tumor_volume)# Find indices where tumor volume exceeds the maximum allowed. welfare_missing = np.where(tumor_volume > max_tumor_volume)[0]iflen(welfare_missing) >1:# Exclude from its first occurrence until the next timepoint for survival . tumor_volume[welfare_missing[1:]] = np.nan# Create a DataFrame for the current animal's data animal_data = pd.DataFrame( {"day": days,"animal": f"{group_name}_{i +1}","group": group_name,"tv": tumor_volume, } )# Add the animal's data to the list of group data group_data.append(animal_data)# Concatenate all animal dataframes into one for the entire group# ignore_index=True resets the index of the resulting DataFrame.return pd.concat(group_data, ignore_index=True)
This next dataset models a preclinical study with:
Four groups
control (vehicle)
therapy_a (single agent)
therapy_b (single agent)
therapy_combo_ab (combination)
Study design
20 animals per group
24-day duration
Tumor volume measurements every 3 days
25% coefficient of variation (CV) for measurement error
Tumor growth dynamics
Initial tumor volume: 100 mm³ for all animals
Growth rate: each group has a different average daily growth rate (5%, 4.5%, 3.5%, 1%
respectively), with 10% CV within each group.
Decay rate: each group (except combo therapy) has a different average daily decay rate (0.2%,
0.125%, 0.1%, 0% respectively), also with 10% CV within each group.
Code
# Set a seed for reproducibilitynp.random.seed(1234)# Simulate the vehicle (control) group datavehicle_group_quad = simulate_quad_exp_growth( group_name="control", duration=24, initial_tumor_volume=100, group_growth_rate_day_pct=5, growth_rate_cv=0.1, group_decay_rate_day_pct=.2, decay_rate_cv=0.1, residual_cv=0.25, n_animals=20, max_tumor_volume=4000)# Simulate data for the single-agent therapy A grouptherapy_a_group_quad = simulate_quad_exp_growth( group_name="therapy_a", duration=24, initial_tumor_volume=100, group_growth_rate_day_pct=4.5, growth_rate_cv=0.1, group_decay_rate_day_pct=0.125, decay_rate_cv=0.1, residual_cv=0.25, n_animals=20, max_tumor_volume=4000)# Simulate data for the single-agent therapy B grouptherapy_b_group_quad = simulate_quad_exp_growth( group_name="therapy_b", duration=24, initial_tumor_volume=100, group_growth_rate_day_pct=3.5, growth_rate_cv=0.1, group_decay_rate_day_pct=0.1, decay_rate_cv=0.1, residual_cv=0.25, n_animals=20, max_tumor_volume=4000)# Simulate data for the combination therapy A+B grouptherapy_ab_group_quad = simulate_quad_exp_growth( group_name="therapy_combo_ab", duration=24, initial_tumor_volume=100, group_growth_rate_day_pct=1, growth_rate_cv=0.1, group_decay_rate_day_pct=0, decay_rate_cv=0.1, residual_cv=0.25, n_animals=20, max_tumor_volume=4000)# Combine all group data into a single DataFrameTGI_dataset_quad = pd.concat( [ vehicle_group_quad, therapy_a_group_quad, therapy_b_group_quad, therapy_ab_group_quad ], ignore_index=True)# Let's have a look at the summary statistics dataset structure and valuesprint(summary_stats_tgi(TGI_dataset_quad).head(18))
# For comparison with the R tutorial we can import the original TGI.datasetTGI_dataset_quad = pd.read_csv("TGI_dataset_quadratic.csv")TGI_dataset_quad.columns = [col.lower() for col in TGI_dataset_quad.columns]for col in TGI_dataset_quad.select_dtypes(include='object').columns: TGI_dataset_quad[col] = TGI_dataset_quad[col].astype(str).str.lower()TGI_dataset.head(6)
To accurately analyze this type of tumor growth, we can employ a quadratic exponential model. This model
incorporates both a tumor growth rate (TGR) and a tumor decay rate (TDR) to capture the
non-linear growth dynamics.
The following function estimates and compares the TGR and TDR of different treatment groups against a
control group or another treatment group. It allows for both one-sided and two-sided hypothesis tests,
providing flexibility for different research questions.
Code
############################################################################## USE THIS FUNCTION WITH CAUTION ! IT DOESN'T GIVE THE SAME RESULTS AS THE ## ORIGINAL R SNIPPET, THOUGH THE R SNIPPET FAILS TO SUMMARIZE PROPERLY THE ## TGR AND TDR VALUES, AS WELL AS THE P VALUES AND THE EXPECTED CONCLUSIONS. ##############################################################################def tumor_growth_curves_quadratic_exponential( dataset: pd.DataFrame, treatments_numerator: List[str], treatment_denominator: str='control', time_points: List[int] =None, p_tails: int=1) -> pd.DataFrame:""" Estimates and compares tumor growth rates (TGR) and tumor decay rates (TDR) between treatment groups using a linear mixed-effects model with a quadratic term for time. Args: dataset (pd.DataFrame): DataFrame with columns 'day', 'group', and 'tv' (tumor volume). treatments_numerator (List[str]): List of treatment group names. treatment_denominator (str, optional): Name of the comparison group. Defaults to 'control'. time_points (List[int], optional): List of days at which to calculate TGR. If None, uses all unique days. p_tails (int, optional): 1 for one-sided test (tumor reduction vs. control), 2 for two-sided test. Defaults to 1. Returns: pd.DataFrame: DataFrame containing TGR and TDR estimates, with their confidence intervals, and P-values for each contrast. """# Filter the dataset for relevant groups and time points included_groups = treatments_numerator + [treatment_denominator]ifnot time_points:# uses all unique days in the dataset time_points = dataset['day'].unique() tv_data = dataset[ (dataset['group'].isin(included_groups))& (dataset['day'].isin(time_points)) ].copy()# Log-transform tumor volume tv_data['log_tv'] = np.log(tv_data['tv'])# Convert 'day' to numeric and 'group' to categorical variable (good practice) tv_data['day'] = pd.to_numeric(tv_data['day']) tv_data['group'] = pd.Categorical( tv_data['group'], categories=[treatment_denominator] + treatments_numerator)# Generate orthogonal polynomial features for Day (degree 2) poly_features = np.polynomial.polynomial.polyvander(tv_data['day'], deg=2) tv_data['day_linear'] = poly_features[:, 1] # Linear term tv_data['day_quadratic'] = poly_features[:, 2] # Quadratic term# Fit a linear mixed-effects model with a quadratic time component and random effects for both intercept and slope model = smf.mixedlm(# equivalent to R formula 'lnTV ~ Group * poly(Day, 2), random = ~ 1 + poly(Day, 2) | Animal'f"log_tv ~ C(group, Treatment(reference='{ treatment_denominator}')) * (day_linear + day_quadratic)", data=tv_data, groups=tv_data["animal"], re_formula="day_linear + day_quadratic", missing='drop') est = model.fit( reml=True, # default method=["lbfgs"] )# Extract model estimates for the interaction terms (group:day)# print(est.summary()) results = est.summary().tables[1]# Need to trick a little bit the outputfor col in results.columns: results[col] = pd.to_numeric(results[col], errors='coerce')# Results container model_estimates_list = []for treatment in treatments_numerator:# Extract TGI, TGR, and their CI, and P value related to the interaction terms with# the quadratic day component ('poly(day, 2)') ix_tgr =f"C(group, Treatment(reference='{treatment_denominator}'))[T.{ treatment}]:day_linear" ix_tdr =f"C(group, Treatment(reference='{treatment_denominator}'))[T.{ treatment}]:day_quadratic" tgr = results.loc[ix_tgr, "Coef."] tdr = results.loc[ix_tdr, "Coef."]# Calculate confidence intervals and P-values for TGR and TDR std_err_tgr = results.loc[ix_tgr, "Std.Err."] std_err_tdr = results.loc[ix_tdr, "Std.Err."]if p_tails ==1: lb95_tgr = np.nan ub95_tgr = results.loc[ix_tgr, "Coef."] +\ norm.ppf(0.95) * std_err_tgr# Although the t-distribution is used in R pvalue_tgr = norm.cdf(results.loc[ix_tgr, "z"]) lb95_tdr = np.nan ub95_tdr = results.loc[ix_tdr, "Coef."] +\ norm.ppf(0.95) * std_err_tdr# Although the t-distribution is used in R pvalue_tdr = norm.cdf(results.loc[ix_tdr, "z"])# we just pickup the results from statsmodels as it (normal distribution)else: lb95_tgr = results.loc[ix_tgr, "[0.025"] ub95_tgr = results.loc[ix_tgr, "0.975]"] pvalue_tgr = results.loc[ix_tgr, "P>|z|"] lb95_tdr = results.loc[ix_tdr, "[0.025"] ub95_tdr = results.loc[ix_tgr, "0.975]"] pvalue_tdr = results.loc[ix_tdr, "P>|z|"]# Create a dataframe of the results for the current treatment model_estimates = pd.DataFrame({"contrast": [f"{treatment} vs. {treatment_denominator}"],"time_window": [f"{tv_data['day'].min()}-{tv_data['day'].max()}"],"tgr": [tgr],"lb95_tgr": [lb95_tgr],"ub95_tgr": [ub95_tgr],"pvalue_tgr": [pvalue_tgr],"tdr": [tdr],"lb95_tdr": [lb95_tdr],"ub95_tdr": [ub95_tdr],"pvalue_tdr": [pvalue_tdr], })# Append results for this treatment to the list model_estimates_list.append(model_estimates)# Combine results into a single DataFramereturn pd.concat(model_estimates_list, ignore_index=True)
The discrepancies in intercept, TGR, and TDR values between R’s lme and Python’s
statsmodels.mixedlm can be attributed to several factors:
Differences in estimation algorithms:
R (lme): the lme function in the nlme package typically uses Restricted
Maximum Likelihood (REML) as the default estimation method for mixed-effects models. REML aims to
provide unbiased estimates of the variance components (the random effects).
Python (statsmodels): The mixedlm function in statsmodels also uses REML
as a default method. However, there might be subtle differences in the specific algorithms and
optimizations used in each library, leading to slightly different estimates.
Differences in default settings:
Convergence criteria: the convergence criteria for the iterative optimization algorithms used to
fit the models might differ between the two libraries. This could lead to slightly different
stopping points and therefore different estimates.
Parameterization of random effects: the way the random effects are parameterized and the default
assumptions about their distribution (e.g., unstructured vs. diagonal covariance matrix) might
also vary slightly, contributing to the differences.
Numerical precision:
The underlying numerical libraries and calculations used by R and Python might have slight
differences in precision, which can accumulate and lead to small variations in the final estimates.
Interpreting the results:
Intercept: the intercept represents the baseline log-transformed tumor volume for the reference group
at day 0. The difference between the R and Python estimates (e.g., 5.622 vs. 4.493) suggests a
slight shift in the baseline level between the two models.
TGR: the TGR represents the average change in log tumor volume per day for each group. The discrepancy
between R (3.8543) and Python (-0.042) is more substantial. This might indicate a difference in how the
models are estimating the time-dependent effect of the treatment.
TDR: the TDR represents the acceleration or deceleration in the growth rate over time. Again, the
difference between R (2.81) and Python (0.003) suggests some variation in how the quadratic term is
being estimated by the two models.
While the exact numerical values might differ, the overall conclusions about the relative effects of the
treatments should be consistent between the two implementations. The statistical significance of the
treatment effects (as indicated by p-values) should also be similar, although minor differences are
possible due to the variations in degrees of freedom and estimation methods.
Remember that the goal is to understand the relative differences between groups, not to obsess over minor
numerical discrepancies. If the qualitative conclusions are similar between R and Python, then we can be
reasonably confident in the findings.
4.2.4.6 Modeling long-term tumor dynamics with the Gompertzian
model
While exponential and quadratic exponential models are useful for capturing tumor growth patterns over
shorter periods, they have a key limitation: they predict unlimited growth. However, long-term experiments
reveal that tumor growth doesn’t continue indefinitely. It eventually plateaus due to factors like limited
resources and space constraints within the body.
The Gompertzian
model addresses this limitation by introducing a theoretical upper limit (\(y_\text{max}\)) to tumor volume. This means that the model predicts tumor
growth will slow down and eventually stop as it approaches this maximum size, more accurately reflecting
real-world observations.
The Gompertzian model is applied to a transformed version of the tumor volume data, i.e., \({y}_{it}^{\dagger }=\ln \left(\ln \left({y}_{max}/{y}_{it}\right)\right)\).
This double log-transformation linearizes the relationship between tumor growth and time. The
model can then be expressed as:
\(E\left({Y}_{it}^{\dagger}\right)\): expected value of the double
log-transformed tumor volume for animal \(i\) at time \(t\).
\({\eta}_j\): a parameter related to the initial tumor volume and
growth rate for treatment group \(j\).
\({\lambda}_j\): the Gompertzian force for treatment
group \(j\), which determines the rate at which tumor growth slows
down.
\({t_{ij}}^{\ast }\): time of measurement for animal \(i\) in group \(j\).
The \({\eta}_j\) parameter combines information about the initial tumor
volume (\(\alpha_j\)) and the initial growth rate (\(\beta_j\)) for group \(j\), i.e., \({\eta}_j=\ln \left({\beta}_j/{\alpha}_j\right)\). A higher \({\eta}_j\) indicates a larger initial tumor or faster initial growth.
\({\lambda}_j\) is the key parameter in the Gompertzian model. It
represents the “force of mortality” or the rate at which the tumor’s growth potential decreases over time.
A larger positive value of \({\lambda}_j\) means the tumor growth slows
down more quickly, and when \({\lambda}_j\) approaches zero, the
Gompertzian model reduces to the exponential growth model, indicating constant growth.
Finally, the growth rate at any given time can be calculated as \({\beta_j}^{\ast}\mathit{\exp}\left({t_{ij}}^{\ast }{\lambda}_j\right)\).
This shows that the growth rate decreases exponentially over time due to the Gompertzian force.
The Gompertzian model requires knowledge of the maximum tumor volume (\(y_\text{max}\)), which can only be estimated accurately by allowing tumors
to grow for extended periods until they reach a plateau. This raises ethical concerns about
animal welfare, as it can lead to prolonged suffering.
The Gompertzian model provides a more realistic representation of tumor growth compared to the
exponential or quadratic exponential models, especially for long-term studies. It incorporates the concept
of a carrying capacity, which represents the maximum tumor volume achievable due to biological
constraints. We’ll simulate data following this model while using log-normal distribution for individual
tumor volumes to introduce realistic variability. In contrast to shorter-term studies where animal welfare
considerations might limit tumor size, the goal of this simulation is to explore the theoretical long-term
dynamics of tumor growth under different treatment conditions. Therefore, we won’t be implementing the
concept of “welfare missing,” as we’re interested in observing how tumor volumes evolve over an extended
period, even if they exceed what would be ethically permissible in a real animal study.
Code
def simulate_gompertz_growth( group_name: str, duration: int, initial_tumor_volume: float, maximum_tumor_volume: float, group_growth_rate_day_pct: float, growth_rate_cv: float, residual_cv: float, n_animals: int, df=None) -> pd.DataFrame:""" Simulates tumor growth data for a group of animals over a given duration, using the Gompertzian model with log-normal error. Args: group_name (str): Name of the group (e.g., "control", "therapy_a"). duration (int): Duration of the study in days. initial_tumor_volume (float): Initial tumor volume (mm^3). maximum_tumor_volume (float): Maximum theoretical tumor volume (mm^3). group_growth_rate_day_pct (float): Average initial daily growth rate of the group (%). growth_rate_cv (float): Coefficient of variation for the growth rate within the group. residual_cv (float): Coefficient of variation for the measurement error. n_animals (int): Number of animals in the group. df (int, optional): Degrees of freedom for the t-distribution (growth rate variation). If None, defaults to n_animals - 1. Returns: pd.DataFrame: Simulated data with columns: day, animal, group, tv (Tumor Volume). """ group_data = []for i inrange(n_animals): days = np.arange(0, duration +1, 3)# Sample individual growth rates from a t-distribution df = n_animals -1if df isNoneelse df# growth.rate.individual <- rnorm(n = (length(days) - 1), mean = group.growth.rate.day.perc / 100,# sd = group.growth.rate.day.perc * growth.rate.cv / 100) growth_rate_individual = t_dist.rvs( df=df, loc=group_growth_rate_day_pct /100, scale=group_growth_rate_day_pct * growth_rate_cv /100, size=len(days) -1 )# Calculate carrying capacity on the log scale (log of maximum tumor volume) carrying_capacity = np.log(maximum_tumor_volume) ln_tumor_volume = np.empty(len(days))# Day 0 simulation using normal distribution# ln.tumor.volume[1] <- rnorm(n = 1, mean = log(initial.tumor.volume), sd = sqrt(log((residual.cv^2) + 1))) ln_tumor_volume[0] = np.random.normal( loc=np.log(initial_tumor_volume), scale=np.sqrt(np.log(residual_cv**2+1)) )# Simulate tumor volumes using the Gompertzian growth equationfor t inrange(1, len(days)):# growth.term <- (ln.tumor.volume[1] - carrying.capacity) * exp(-growth.rate.individual[t - 1] * days[t])# ln.tumor.volume[t] <- rnorm(n = 1, mean = carrying.capacity + growth.term, sd = sqrt(log((residual.cv^2) + 1)))# Calculate mean and standard deviation of the underlying normal distribution for log-normal growth_term = (ln_tumor_volume[0] - carrying_capacity) *\ np.exp(-growth_rate_individual[t-1] * days[t]) ln_tumor_volume[t] = np.random.normal( loc=carrying_capacity + growth_term, scale=np.sqrt(np.log(residual_cv**2+1)), )# Store the data for the current animal in a DataFrame animal_data = pd.DataFrame({"day": days,"animal": f"{group_name}_{i +1}","group": group_name,# Retrieve the tumor volume in linear scale"tv": np.exp(ln_tumor_volume), }) group_data.append(animal_data)# Combine all animal dataframes into one for the entire group.return pd.concat(group_data, ignore_index=True)
The simulated dataset will be created with the following characteristics:
Four groups:
Control (vehicle)
therapy_a (single agent)
therapy_b (single agent)
therapy_combo_ab (combination)
Study design:
20 animals per group
60-day duration
Tumor volume measurements every 3 days
25% coefficient of variation (CV) for measurement error
Tumor growth dynamics:
Initial tumor volume: 100 mm³ for all animals
Maximum tumor volume: 6000 mm³ (theoretical limit)
Growth rate: Each group has a different average daily growth rate (6%, 4%, 3%, 2% respectively),
with 10% CV within each group
Code
# Set random seed for reproducibilitynp.random.seed(1)# Simulate the control group (Vehicle) datacontrol = simulate_gompertz_growth( group_name="control", duration=60, initial_tumor_volume=100, maximum_tumor_volume=6000, group_growth_rate_day_pct=6, growth_rate_cv=0.1, residual_cv=0.25, n_animals=20)# Simulate data for Therapy_A grouptherapy_a = simulate_gompertz_growth( group_name="therapy_a", duration=60, initial_tumor_volume=100, maximum_tumor_volume=6000, group_growth_rate_day_pct=4, growth_rate_cv=0.1, residual_cv=0.25, n_animals=20)# Simulate data for Therapy_B grouptherapy_b = simulate_gompertz_growth( group_name="therapy_b", duration=60, initial_tumor_volume=100, maximum_tumor_volume=6000, group_growth_rate_day_pct=3, growth_rate_cv=0.1, residual_cv=0.25, n_animals=20)# Simulate data for Therapy_Combo_AB grouptherapy_combo_ab = simulate_gompertz_growth( group_name="therapy_combo_ab", duration=60, initial_tumor_volume=100, maximum_tumor_volume=6000, group_growth_rate_day_pct=2, growth_rate_cv=0.1, residual_cv=0.25, n_animals=20)# Combine all group data into a single DataFrameTGI_dataset_gompertz = pd.concat( [control, therapy_a, therapy_b, therapy_combo_ab], ignore_index=True)TGI_dataset_gompertz.head(6)
# For comparison with the R tutorial we can import the original TGI.datasetTGI_dataset_gompertz = pd.read_csv("TGI_dataset_gompertz.csv")TGI_dataset_gompertz.columns = [ col.lower() for col in TGI_dataset_gompertz.columns]for col in TGI_dataset_gompertz.select_dtypes(include='object').columns: TGI_dataset_gompertz[col] = TGI_dataset_gompertz[col].astype(str).str.lower()TGI_dataset_gompertz.head(6)
day
animal
group
tv
0
0
control_1
control
115.507965
1
3
control_1
control
330.419462
2
6
control_1
control
321.841189
3
9
control_1
control
386.864409
4
12
control_1
control
642.549477
5
15
control_1
control
1411.567508
4.2.4.7 Long-term TGI studies and the Gompertzian model
As we discussed earlier, the Gompertzian model is well-suited for analyzing long-term tumor growth
inhibition (TGI) studies where tumors might reach a plateau due to factors like limited resources or
space. In this model, we transform the tumor volume data using the following equation \({y}_{ij k}^{\dagger }=\ln \left(\ln \left({y}_{max}/{y}_{ij
k}\right)\right)\), where \({y}_{max}\) is the maximum tumor
volume (carrying capacity), and \({y}_{ij k}\) is the observed tumor
volume for animal \(i\) in group \(j\)
at time \(k\). This transformation makes the relationship between the
transformed tumor volume (\({y}_{ij k}\)) and time approximately linear,
allowing us to use a linear mixed-effects model for analysis.
The following formula defines a mixed-effects model to analyze tumor growth rates (TGRs) in a
longitudinal TGI study, assuming that the tumor growth follows a Gompertzian pattern:
\(E\left({y}_{ij k}^{\dagger}\right)\): this represents the
expected value (average) of the transformed tumor volume (\({y}_{ij
k}^{\dagger}\)) for animal \(i\) in group \(j\) at the \(k\)-th time point. The
dagger (\(^{\dagger}\)) symbol indicates the transformation of the
original tumor volume (\(y\)) using the Gompertzian formula.
\({\eta}_j\): this is the fixed effect for the intercept of the \(j\)-th group. It represents the average initial transformed tumor volume
for animals in group \(j\) at the beginning of the study (\(t=0\)).
\({n}_{ij}\): this is the random effect for the intercept, capturing
individual variations in the initial transformed tumor volume among animals within the same group. It’s
assumed to be normally distributed with a mean of 0.
\({\lambda}_j\): this is the fixed effect for the slope of the \(j\)-th group. It represents the average rate of change of the transformed
tumor volume over time for animals in group \(j\) (the Gompertzian
force).
\({l}_{ij}\): this is the random effect for the slope, accounting for
individual variations in the tumor growth rate among animals within the same group. It’s assumed to be
normally distributed with a mean of 0.
\({t}_{ij k}\): this is the time point of the \(k\)-th measurement for animal \(i\) in
group \(j\).
How to interpret these terms?
Fixed effects (\({\eta}_j\), \({\lambda}_j\)): these parameters capture the average initial transformed
tumor volume and the average growth rate for each treatment group, respectively. By comparing these
estimates across groups, we can assess the effects of different treatments on tumor growth dynamics.
Random effects (\({n}_{ij}\), \({l}_{ij}\)): these parameters account for the variability in the initial
tumor volume and growth rate among individual animals within each group. The mixed-effects model
framework allows the estimatation of these random effects and their variance components, providing a
more comprehensive understanding of the variability in tumor growth patterns.
We can implement this model in Python using the statsmodels.formula.api.mixedlm function,
similar to how we did for the exponential model. The formula would be adapted to use the
Gompertz-transformed outcome variable and the random effects structure.
Code
def tumor_growth_curves_gompertzian( dataset: pd.DataFrame, treatments_numerator: List[str], treatment_denominator: str='control', p_tails: int=1) -> pd.DataFrame:""" Estimates and compares Gompertzian parameters (lambda) between treatment groups using a linear mixed-effects model. Args: dataset (pd.DataFrame): DataFrame with columns 'day', 'group', 'animal', and 'tv' (tumor volume). treatments_numerator (List[str]): List of treatment group names (numerator in the comparison). treatment_denominator (str, optional): Name of the comparison group (denominator). Defaults to 'control'. p_tails (int, optional): 1 for one-sided test (tumor reduction), 2 for two-sided test. Defaults to 1. Returns: pd.DataFrame: DataFrame containing Gompertzian parameter estimates, confidence intervals, and p-values for each contrast. """# Filter the dataset for relevant groups and time points included_groups = treatments_numerator + [treatment_denominator] tv_data = dataset[dataset['group'].isin(included_groups)].copy()# Create Gompertz transformed variable# Pseudocount to avoid division by zero espilon =1e-8 tv_data["y_cross"] = np.log( np.log(tv_data['tv'].max() / (tv_data['tv'] + epsilon) + epsilon))# Handle undefined values (-inf) by replacing them with a very small negative number# tv_data["y_cross"] = tv_data["y_cross"].replace(-np.inf, tv_data["y_cross"][np.isfinite(tv_data["y_cross"])].min()) tv_data["y_cross"] = tv_data["y_cross"].replace(-np.inf, espilon)# Ensure 'day' is numeric and convert 'group' to categorical variable (good practice) tv_data['day'] = pd.to_numeric(tv_data['day']) tv_data['group'] = pd.Categorical( tv_data['group'], categories=[treatment_denominator] + treatments_numerator)# Fit a linear mixed-effects model model = smf.mixedlm(# equivalent to R formula 'y.cross ~ Group * Day, random = ~ 1 + Day | Animal'f"y_cross ~ C(group, Treatment(reference='{ treatment_denominator}')) * day", data=tv_data, groups=tv_data["animal"], re_formula="day", missing='drop') est = model.fit( reml=True, # default# method=["lbfgs"] )# Extract model estimates for the interaction terms (group:day)# print(est.summary()) results = est.summary().tables[1]# Need to trick a little bit the outputfor col in results.columns: results[col] = pd.to_numeric(results[col], errors='coerce')# Results container model_estimates_list = []for treatment in treatments_numerator:# Extract model estimates ix =f"C(group, Treatment(reference='{ treatment_denominator}'))[T.{treatment}]:day" Gompertz_par = results.loc[ix, "Coef."]# Calculate confidence intervals and P-values for TGR and TDR std_err = results.loc[ix, "Std.Err."]if p_tails ==1: lb95 = np.nan ub95 = Gompertz_par + norm.ppf(0.95) * std_err# Although the t-distribution is used in R pvalue =1- norm.cdf(results.loc[ix, "z"])# we just pickup the results from statsmodels as it (normal distribution)else: lb95 = results.loc[ix, "[0.025"] ub95 = results.loc[ix, "0.975]"] pvalue = results.loc[ix, "P>|z|"]# Create a dataframe of the results for the current treatment model_estimates = pd.DataFrame({"contrast": [f"{treatment} vs. {treatment_denominator}"],"Gompertz_par": [Gompertz_par],"lb95": [lb95],"ub95": [ub95],"pvalue": [pvalue], })# Append results for this treatment to the list model_estimates_list.append(model_estimates)# Combine results into a single DataFramereturn pd.concat(model_estimates_list, ignore_index=True)
The Gompertzian parameter (often denoted as \(\lambda\)) represents the
rate at which tumor growth slows down over time. A positive ‘Gompertz.par’ of 0.01 indicates that
‘therapy_a’ is associated with a slightly higher rate of tumor growth deceleration compared to the control
group. In other words, tumors in the ‘therapy_a’ group are slowing down in their growth rate a bit faster
than those in the control group.
The magnitude of 0.01 suggests that the difference in deceleration rates is relatively small. It’s
essential to interpret this in the context of the specific units of time used in the study (e.g., days,
weeks) and the overall range of Gompertzian parameters observed in the data. The interpretation also
depends on whether this difference is statistically significant. If the associated P value is small (e.g.,
less than 0.05), it suggests that the observed difference is unlikely to be due to chance alone, and
‘therapy_a’ likely has a true effect on tumor growth deceleration.
The ‘Gompertz.par’ value alone doesn’t tell us the absolute growth rates or tumor sizes in each group. It
only represents the difference in the rate of change of the growth rates. To fully understand the impact
of the treatment, we would also need to look at the estimated TGR (Tumor Growth Rate) and the actual tumor
volumes over time for both groups. The biological interpretation of the Gompertzian force depends on the
specific context of the study and the tumor type we’re investigating.
4.2.4.8 Summarizing tumor growth with AUC
When analyzing longitudinal tumor growth data, one way to condense the information from multiple time
points is to calculate the Area Under the Curve (AUC) for each animal’s growth
trajectory. The AUC represents the cumulative effect of tumor growth over time, providing a single value
for each animal that can be used for statistical comparisons between groups.
Why use log-transformed values (\({y}_{ijt}^{\ast }\))?
Balancing early and late time points: in tumor growth curves, early time points often show
rapid growth, while later time points may show slower growth or even a plateau. Using the raw tumor
volumes (\({y}_{ijt}^{\ast }\)) for AUC calculation can give undue
weight to these early rapid growth phases. Log-transforming the values (\({y}_{ijt}^{\ast }\) = ln({y}_{ijt}))$ helps to balance the influence of
early and late time points, ensuring a more even contribution across the study period.
Standardizing to zero: subtracting the initial log-transformed tumor volume (\({y}_{ij1}^{\ast }\)) from all subsequent values for the same animal
standardizes the growth curves to start at zero. This focuses the AUC analysis on the change in tumor
growth over time rather than absolute tumor volumes, which can vary between animals even within the same
group.
While various methods exist for calculating AUC, the trapezoidal rule is the most
commonly used due to its simplicity and ease of implementation. It approximates the area under the curve
by dividing it into trapezoids and summing their areas, with \(\text{AUC}_{t_1 -
t_2} \approx \sum[1/2 * ({y}_{ijt_1}^{\ast } + {y}_{ijt_2}^{\ast }) * (t_2 - t_1)]\).
Once we have calculated the AUC values for each animal, you can treat these values as your outcome
variable and perform standard statistical tests like t-tests (for two groups) or ANOVA (for multiple
groups) to compare the average AUC between treatment and control groups. This tells us if there’s a
statistically significant difference in the overall tumor growth pattern over time.
4.2.4.9 AUC analysis with Python
In addition to examining specific time points or growth rates, analyzing the area under the curve (AUC)
of tumor growth curves provides a holistic view of the overall tumor burden over time. The AUC summarizes
the cumulative effect of treatment across the entire study period, offering a complementary perspective to
TGR analysis.
The following function, tumor_auc, calculates and compares the AUC of different treatment
groups, providing a comprehensive assessment of treatment efficacy in inhibiting tumor growth.
Code
from scipy.integrate import trapezoid as trapz# https://docs.scipy.org/doc/scipy/reference/generated/scipy.integrate.trapezoid.htmldef tumor_auc( dataset: pd.DataFrame, treatments_numerator: List[str], treatment_denominator: str='control', p_tails: int=1) -> pd.DataFrame:""" Calculates and compares area under the curve (AUC) of tumor growth curves between treatment groups. Args: dataset (pd.DataFrame): DataFrame with columns 'day', 'group', 'animal', and 'tv' (tumor volume). treatments_numerator (List[str]): List of treatment group names. treatment_denominator (str, optional): Name of the comparison group. Defaults to 'control'. p_tails (int, optional): 1 for one-sided test (tumor reduction), 2 for two-sided test. Defaults to 1. Returns: pd.DataFrame: DataFrame with columns 'contrast', 'AUC_diff', 'lb95_AUC_diff', 'ub95_AUC_diff', and 'pvalue'. """# Filter relevant data tv_data = dataset[dataset['group'].isin( treatments_numerator + [treatment_denominator])].copy()# Log-transform tumor volume (tv) and add log_tv column tv_data['log_tv'] = np.log(tv_data['tv'])# Extract baseline (first day) data tv_data['log_tv_baseline'] = tv_data.groupby('animal')['log_tv'].transform('first')# remove baseline from further calculations for standardization, to avoid bias,# and focusing on treatment's effect on tumor growth dynamics tv_data = tv_data.query('day != day.min()').copy()# Calculate height for AUC (subtracting baseline and setting negative values to 0) tv_data['height'] = tv_data['log_tv'] - tv_data['log_tv_baseline'] tv_data['height'] = tv_data['height'].apply(lambda x: max(0, x)) # Ensure non-negative heights# Calculate AUC for each animal using trapezoidal ruledef calculate_auc(x): x = x.sort_values('day')return trapz(x['height'], x['day']) auc_data = ( tv_data .groupby('animal') .apply( calculate_auc, include_groups=False ) .reset_index(name='AUC') )# Merge AUC data with original data to get group information auc_data = auc_data.merge( tv_data[['animal', 'group']], on='animal', how='left').drop_duplicates()# Convert group to factor auc_data['group'] = pd.Categorical( auc_data['group'], categories=[treatment_denominator] + treatments_numerator)# Fit a linear model to compare AUCs between groups model = smf.ols('AUC ~ group', data=auc_data) est = model.fit() degrees_of_freedom = est.df_resid# Extract model estimates for the interaction terms (group:day)# print(est.summary2()) results = est.summary2().tables[1]# Results container model_estimates_list = []for treatment in treatments_numerator:# Extract model estimates ix =f"group[T.{treatment}]" Gompertz_par = results.loc[ix, "Coef."]# Calculate confidence intervals and P-values for TGR and TDR std_err = results.loc[ix, "Std.Err."]if p_tails ==1: lb95 = np.nan ub95 = Gompertz_par +t_dist.ppf(0.95, df=degrees_of_freedom) * std_err pvalue = t_dist.cdf(results.loc[ix, "t"], df=degrees_of_freedom)# we just pickup the results from statsmodels as it (normal distribution)else: lb95 = results.loc[ix, "[0.025"] ub95 = results.loc[ix, "0.975]"] pvalue = results.loc[ix, "P>|t|"]# Create a dataframe of the results for the current treatment model_estimates = pd.DataFrame({"contrast": [f"{treatment} vs. {treatment_denominator}"],"AUC_diff": [Gompertz_par],"lb95_AUC_diff": [lb95],"ub95_AUC_diff": [ub95],"pvalue": [pvalue], })# Append results for this treatment to the list model_estimates_list.append(model_estimates)# Combine results into a single DataFramereturn pd.concat(model_estimates_list, ignore_index=True)
Let’s first call tumor_auc to compare the overall tumor burden (AUC) of three treatment
groups (‘therapy_a’, ‘therapy_b’, and ‘therapy_combo_ab’) to the control group across the entire study
duration. The goal is to determine if any of the treatments lead to a statistically significant reduction
in tumor burden compared to the control.
The ‘AUC_diff’ value is the estimated difference in mean AUC between the treatment group and the control
group. A negative value indicates that the treatment groups have a lower AUC, suggesting a reduction in
tumor burden.
Let’s now call aims the function to directly compare the AUC between two treatment groups (‘therapy_a’
and ‘therapy_b’) to assess if one is significantly more effective than the other in reducing tumor burden.
The very high P value, well above the commonly used significance level of 0.05, indicates that we do not
have sufficient statistical evidence to reject the null hypothesis. In this context, the null hypothesis
is that there is no difference in the mean AUC between ‘therapy_a’ and ‘therapy_b’.
4.2.5 Linear models: the workhorses of longitudinal TGI analysis
The exponential, quadratic exponential, and Gompertzian models, despite their different assumptions about
growth patterns, all share the advantage of being linear models when the tumor volumes are appropriately
transformed (logarithmic or Gompertz transformation). This linearity makes them easier to compute and
interpret compared to more complex non-linear models.
While piecewise linear models (not discussed here) offer another way to handle changes in growth rates over
time, they might be less intuitive to interpret and require more complex model specification. Non-linear
models, although potentially more flexible, are computationally challenging and prone to convergence issues,
especially with noisy data or sparse measurements typical in TGI studies. Additionally, rank-based
non-parametric methods exist for longitudinal data, but they are not commonly used in this context and are
not covered here.
The emphasis is on the practical application of the linear models presented. They provide
a good balance between capturing the essential dynamics of tumor growth and being computationally tractable
for most TGI studies. This prioritizes ease of use and interpretation for researchers without sacrificing
too much accuracy
4.3 Time-to-event analysis in preclinical cancer studies
Time-to-event (TTE) analysis is used to estimate how long it takes for a specific event
(e.g., tumor reaching a certain size, animal death) to occur under different treatment conditions.
Event definition: the event of interest can be defined in various ways, depending on the study goals and
the biology of the tumor.
Censored observations: animals that don’t experience the event by the end of the study, or are removed for
other reasons, are considered censored. This means we know they didn’t have the event up to
a certain point, but we don’t know when or if it would have happened later.
Assumption of independent censoring: TTE analysis typically assumes that censoring is independent of
prognosis, meaning the reasons for censoring (e.g., end of study) are unrelated to whether or not the animal
would have eventually experienced the event. This assumption is often reasonable in TGI studies.
Statistical methods:
Kaplan-Meier curves: used to estimate the survival (or event-free) probability over
time.
Log-rank test: used to compare the survival curves of different treatment groups.
Cox proportional hazards model: used to assess the effect of treatment and other factors on the risk
of the event occurring, while accounting for censored observations.
Purpose: the K-M estimator is a non-parametric method used to estimate the survival function, which
tells us the probability of an animal remaining event-free at different time points.
It’s calculated by summing up the number of events that occurred up to time \(t\), where \(Z_i(t)\) is \(1\) if the event happened, \(0\)
otherwise, and dividing by the total number of animals (\(N\)).
Benefits:
Provides a different perspective on TTE data compared to survival curves.
Useful for comparing event rates between groups.
4.3.2 The Cox Proportional Hazards (CPH) model and beyond
When analyzing the time it takes for a specific event (e.g., tumor reaching a certain size) to occur in
preclinical cancer studies, the Cox Proportional Hazards (CPH) model is often preferred over other methods.
More information about the Cox
proportional hazards model and analysis in Python in this jupyter-book.
Why Choose the CPH Model?
Semi-parametric flexibility: the CPH model doesn’t require you to assume a specific distribution for the
survival times. It’s more flexible than parametric models (like Weibull or Accelerated Failure Time) that
make stronger assumptions about the underlying data.
Hazard ratios: the model quantifies the difference in risk (hazard) of the event
occurring between treatment groups using hazard ratios (HR). An HR greater than 1 means
the treatment group has a higher risk, while an HR less than 1 means a lower risk compared to the control.
Covariate adjustment: the CPH model allows to adjust for other factors (covariates) that might influence
the time-to-event, providing a more nuanced analysis of the treatment effect.
Robustness: despite its assumption of proportional hazards (meaning the relative risk between groups
remains constant over time), the CPH model is often quite robust to violations of this assumption.
The hazard function \(\lambda(t)\) represents the instantaneous risk of
the event occurring at time \(t\).
The CPH model expresses the hazard function for each animal as a function of:
Baseline hazard (\(\lambda_0(t)\)): the underlying risk of the event at
time \(t\), common to all animals.
Treatment effects (\(\beta\)): the coefficients that quantify the
increase or decrease in risk associated with each treatment group compared to the control group.
Indicator variables (\(I\)): binary variables (\(0\) or \(1\)) indicating which treatment
group each animal belongs to.
Alternatives to the CPH Model:
Parametric models: these models (like Weibull
or Accelerated Failure Time) make specific assumptions about the distribution of survival times.
They can be more powerful than the CPH model if the assumptions are met, but less robust if the
assumptions are violated.
Log-rank test: the
log-rank test is a non-parametric hypothesis test that compares the survival distributions of two or
more groups. It’s designed to determine whether there are statistically significant differences in the
time it takes for an event of interest to occur between these groups. It’s simpler than the CPH model but
doesn’t allow for covariate adjustment or estimation of hazard ratios. The log-rank test is often used in
TGI studies to compare the time it takes for tumors to reach a certain size (the “event”) under different
treatment conditions. It’s a valuable tool for assessing the long-term efficacy of treatments and can
complement analyses based on tumor growth rates or AUC, but it can also be less powerful than the CPH
model. How it works:
Observed and expected events: at each time point where an event occurs, the log-rank test
calculates:
The observed number of events in each group.
The expected number of events in each group, assuming the null hypothesis is true (no difference
between groups).
Test statistic: the test statistic (\(D\)) is calculated as the sum
of the squared differences between observed and expected events across all time points, weighted by
the number of individuals at risk at each time point.
Distribution of test statistic: under the null hypothesis, the test statistic (\(D\)) approximately follows a chi-squared distribution
with degrees of freedom equal to the number of groups minus one. And the standardized log-rank test
statistics approximately follows a standard normal distribution (\(D/\sqrt{Var(D)}\)) under the null hypothesis. This allows us to use the
standard normal distribution to calculate P-values and assess the statistical significance of the
observed difference between groups.
P value: it is calculated based on the test statistic and its distribution under the null
hypothesis. A small P value (e.g., less than 0.05) indicates that the observed differences between
groups are unlikely to have occurred by chance alone, providing evidence to reject the null
hypothesis.
Fleming-Harrington tests: these are also non-parametric tests, but they can be adjusted to emphasize
early or late differences in hazard rates between groups.
4.3.3 Using Python for TTE analysis in TGI studies
LTTE analysis can reveal differences in the time to tumor progression or the overall survival of animals in
different treatment groups. The following function, tte_analysis, offers two methods for TTE
analysis:
CPH regression: this semi-parametric model estimates hazard ratios, which quantify the relative risk of
an event occurring in different groups over time.
Log-rank test: this non-parametric test compares the survival curves of different groups to determine if
there are statistically significant differences in survival times.
The function also allows for plotting Kaplan-Meier curves, a visual representation of the survival function
over time, to aid in the interpretation of the results.
Code
from lifelines import KaplanMeierFitter, CoxPHFitterfrom lifelines.statistics import logrank_testdef tte_analysis( dataset: pd.DataFrame, event_threshold: float, treatments_numerator: List[str], treatment_denominator: str='control', method: str='cox', plotKM: bool=False, colormap: str="viridis") -> pd.DataFrame:"""Performs time-to-event analysis using either CoxPH regression or log-rank test. Args: dataset (pd.DataFrame): DataFrame with columns 'day', 'group', 'animal', and 'tv' (tumor volume). event_threshold (float): Tumor volume threshold defining the event of interest. treatments_numerator (List[str]): List of treatment group names. treatment_denominator (str, optional): Name of the comparison group. Defaults to 'control'. method (str, optional): Method for TTE analysis ('cox' or 'logrank'). Defaults to 'cox'. plotKM (bool, optional): Whether to plot Kaplan-Meier curves. Defaults to False. colormap (str, optional): Colormap to use for group colors. Defaults to "viridis". Returns: pd.DataFrame: DataFrame containing results of the analysis. """# Filter for relevant groups included_groups = treatments_numerator + [treatment_denominator] tv_data = dataset[dataset['group'].isin(included_groups)].copy()# Ensure 'day' is numeric and convert 'group' to categorical variable (good practice) tv_data['day'] = pd.to_numeric(tv_data['day']) tv_data['group'] = pd.Categorical( tv_data['group'], categories=[treatment_denominator] + treatments_numerator)# The goal is to produce a table compatible with lifelines, such as we can pick up# the time of the first occurence of tumor volume exceeding the threshold ('event'),# or the latest study day if this never happens ('censoring'), e.g.,# 0 3 6 9 12 15 18# animal_a - - - - - x x# ^# animal_b - - - - - - -# ^# Create a Series of observed 'events' based on tumor volume threshold event = ( tv_data .groupby('animal') .tv .apply(lambda x: (x > event_threshold).any()) .rename('event') )# Create a Series of duration to event/censoring for each animal duration = ( tv_data .set_index('day') # for getting the index via idxmax .groupby('animal') # for each animal .tv # working with tumor volumes .apply( # if event detected, take the first datelambda x: (x > event_threshold).idxmax() - tv_data['day'].min()# in the case any event is in the Seriesif (x > event_threshold).any()# otherwise take the last dateelse tv_data['day'].max() - tv_data['day'].min() ) .rename('duration') )# Create lifelines dataFrame using both Series and group information tte_data = pd.merge(left=duration, right=event, on='animal', how='right').merge( tv_data.groupby("animal")["group"].first(), on='animal')# Results container model_estimates_list = []match method:# Cox Proportional Hazards Modelcase'cox':# Fit the Cox model cph = CoxPHFitter()# Fit cph to data cph.fit( df=tte_data, duration_col="duration", event_col="event", formula='group', )# We pick up the columns of interest from the results table results = cph.summary[['coef', 'exp(coef)', 'se(coef)', 'z', 'p', 'exp(coef) lower 95%', 'exp(coef) upper 95%']] results = results.rename( columns={'covariate': 'contrast','exp(coef)': 'hr','exp(coef) lower 95%': 'lb95_hr','exp(coef) upper 95%': 'up95_hr','p': 'pvalue' }) results.insert(0, "contrast", results.index.str.replace(r'^group\[T\.([^\]]+)\]$', rf'\1 vs. {treatment_denominator}', regex=True))# r'^group\[T\.|\]$', '', regex=True) + f" vs. {treatment_denominator}") model_estimates_list.append(results)# Differences in underlying numerical libraries and floating-point arithmetic between# R and Python can lead to minor variations in the calculations and results.# Log-Rank Testcase"logrank":# For each pair of treatment testedfor group in treatments_numerator: ix = tte_data['group'] == group# We pick up the columns of interest from the results table results = logrank_test( durations_A=tte_data[ix]['duration'], durations_B=tte_data[tte_data['group']== treatment_denominator]['duration'], event_observed_A=tte_data[ix]['event'], event_observed_B=tte_data[tte_data['group']== treatment_denominator]['event'], alpha=.95).summary[['test_statistic', 'p']] results = results.rename( columns={'test_statistic': 'chisq','p': 'pvalue' }) results.insert(0, "contrast", f"{group} vs. { treatment_denominator}") model_estimates_list.append(results)case _:raiseValueError("Invalid method. Choose 'cox' or 'logrank'.")# Plot Kaplan-Meier Curves (if requested)if plotKM:# Set up figure fig, ax = plt.subplots(nrows=1, ncols=1, figsize=(6, 5))# Map groups to colors from the colormap, and markers num_groups =len(groups := tte_data['group'].unique()) colors = plt.get_cmap(colormap)(np.linspace(0.25, 1, num_groups))# Create KaplanMeierFitter object kmf = KaplanMeierFitter()for i, group inenumerate(groups): kmf = KaplanMeierFitter() ix = tte_data['group'] == group# Fit the data to the model kmf.fit( durations=tte_data[ix]['duration'], event_observed=tte_data[ix]['event'], label=group) kmf.plot( show_censors=True, ci_show=False, color=colors[i], lw=3, ax=ax)# Formatting ax.set_xlabel("Days", fontsize=12) ax.set_ylabel(r"$1 - \text{P(event)}$", fontsize=12) ax.set_yticks([0, .25, .5, .75, 1]) ax.set_title("Kaplan-Meier analysis", fontsize=14) ax.grid(which='major', axis='y', linestyle='--') ax.legend(loc='lower left', fontsize=10)# Return (combined) resultsreturn pd.concat(model_estimates_list, ignore_index=True)
Time-to-event analysis provides a powerful framework for assessing the efficacy of anticancer treatments by
examining the time it takes for specific events to occur. In the context of TGI studies, this often involves
analyzing the time until a tumor reaches a certain size or progresses to a more advanced stage. The
Kaplan-Meier estimator and the Cox Proportional Hazards model are essential tools in this analysis, offering
both visual and quantitative assessments of treatment effects.
While the methods discussed here are widely applicable, it’s important to be mindful of their limitations,
especially in preclinical studies where sample sizes are often small. The results should be interpreted with
caution, considering the specific context of the study, the choice of statistical method, and the potential
for deviations from the model’s assumptions.
4.4 Analysis of categorical endpoints in TGI studies
In addition to continuous measures like tumor volume, researchers may use categorical endpoints to assess the
efficacy of anticancer treatments in preclinical studies. RECIST (Response Evaluation Criteria in Solid
Tumors) is a widely used system in clinical oncology to categorize patient responses based on changes in tumor
size. TGI studies can adopt a similar approach, classifying animal responses into categories like progressive
disease (PD), stable disease (SD), partial response (PR), and complete response (CR), based on the percentage
change in tumor volume.
The goal is to compare the frequencies of animals in each response category between different treatment
groups:
Two response categories: when there are only two categories (e.g., response vs. no
response), Fisher’s
exact test is appropriate. It’s especially valuable for small sample sizes where the
chi-squared approximation might not be accurate.
Multiple response categories:
Unordered categories: Chi-squared
tests for independence can be used when the categories have no inherent order.
Ordered categories: Cochran-Armitage
test for trend is often preferred when there is a natural order in the
response categories (e.g., PD < SD < PR < CR). This test allows for assigning different weights
to each category, emphasizing the direction of the trend.
The following function, response_analysis, enables us to analyze the frequency distribution of
animals across these response classes at specific time points and compare them between treatment groups using
appropriate statistical tests.
Code
from scipy.stats import fisher_exactfrom statsmodels.stats.contingency_tables import Tabledef response_analysis( dataset: pd.DataFrame, treatments_numerator: str, treatment_denominator: str, day_fin: int, day_ini: int=0, pd_cutoff: float=0.5, pr_cutoff: float=0.3, sd_include: str='non-responder', p_tails: int=2 ) ->tuple[pd.DataFrame, pd.DataFrame]:"""Analyzes categorical response data from a TGI study using RECIST-like criteria. Args: dataset (pd.DataFrame): DataFrame with columns 'day', 'group', 'animal', and 'tv' (tumor volume). treatments_numerator (List[str]): List of treatment group names. treatment_denominator (str): Name of the control (vehicle) group. day_fin (int): Day (final) for assessing response. day_ini (int): Baseline/initial day for tumor volume measurement. pd_cutoff (float, optional): Threshold for Progressive Disease (%). Defaults to 0.5. pr_cutoff (float, optional): Threshold for Partial Response (%). Defaults to 0.3. sd_include (str, optional): How to handle stable disease ('non-responder', 'responder', or 'exclude'). Defaults to 'non-responder'. p_tails (int, optional): 1 for one-sided, 2 for two-sided test. Defaults to 2. Returns: Tuple[pd.DataFrame, pd.DataFrame]: A tuple containing three DataFrames: - The first DataFrame include a 4-level (PD, SD, PR, CR) + binary (responder, non-responder) response representation of the data. - The second DataFrame contains p-values from statistical tests (Cochran-Armitage trend and Fisher's exact). Additional tests, such as the chi-squared test for independence, can be performed using the first DataFrame returned. """# Filter for relevant groups included_groups = treatments_numerator + [treatment_denominator] tv_data = dataset[dataset['group'].isin(included_groups)].copy()# Ensure 'day' is numeric and convert 'group' to categorical variable tv_data['day'] = pd.to_numeric(tv_data['day']) tv_data['group'] = pd.Categorical( tv_data['group'], categories=[treatment_denominator] + treatments_numerator)# Filter data for the specified initial and final days filtered_data = tv_data[ (tv_data['group'].isin(included_groups))& (tv_data['day'].isin([day_ini, day_fin])) ].copy()# Calculate the relative change in tumor volume from day_ini to day_fin response_data = filtered_data.pivot( index=['animal', 'group'], columns='day', values='tv') response_data['relative_change'] = response_data.loc[:, day_fin] / response_data.loc[:, day_ini]# Categorize responses using RECIST-like criteria response_data['response'] = pd.cut( response_data['relative_change'], bins=[-np.inf, 1-pr_cutoff, 1+pd_cutoff, np.inf], labels=['pr', 'sd', 'pd'], right=False# Include lower bound, exclude upper bound )# If the final tumor volume is <= 10, then it's categorized as CR response_data['response'] = response_data['response'].cat.add_categories('cr') ix = response_data[day_fin] <=10 response_data.loc[ix, 'response'] ='cr' response_data['response'] = response_data['response'].cat.reorder_categories( ['pd', 'sd', 'pr', 'cr'])# Create a dataFrame with both response Series and group information response = response_data.reset_index(level='group').loc[:, ['group', 'response']]# Create separate columns for 'responder' and 'non_responder' based on SD_includematch sd_include:case'non-responder': response['responder'] = ( response['response'].isin(['pr', 'cr'])) response['non_responder'] = ( response['response'].isin(['pd', 'sd']))case'responder': response['responder'] = ( response['response'].isin(['pr', 'sd', 'cr'])) response['non_responder'] = ( response['response'] =='pd')case'exclude': response['responder'] = ( response['response'].isin(['pr', 'cr'])) response['non_responder'] = ( response['response'] =='pd')case _:raiseValueError("Invalid sd_include value. Choose from 'non-responder', 'responder', or 'exclude'.")# We drop rows with neither responder or non_responder response = response.drop(response[~response['responder'] &~response['non_responder']].index)# Remove the column name generated from the pivot response.columns.name =''# Print the 4-level and binary contingency tablesprint("Contingency table (quaternary/4-level response):")print( rt4:=pd.crosstab( response['group'], response['response'], dropna=False# Include all categories even with zero counts ) )print("\nContingency table (binary response):")print( rt2:=pd.crosstab( response['group'], response['responder'], dropna=False) )# Initialize lists to store p-values (initialize with NaN to handle cases where the test is not performed) resp_pv = np.full(len(treatments_numerator), np.nan) p_fisher = np.full(len(treatments_numerator), np.nan)# Iterate over treatment groups (start from 0 since enumerate starts from 0)for i, treatment inenumerate(treatments_numerator):# Check if there are enough non-zero cells in the contingency table control_row = rt4.loc[treatment_denominator] treatment_row = rt4.loc[treatment]# Check for at least two non-zero column sumsif np.sum((control_row + treatment_row) !=0) >=2:# Perform Cochran-Armitage Test result = Table( rt4.loc[[treatment_denominator, treatment]], shift_zeros=False ).test_ordinal_association( col_scores=np.array([1, 2, 3, 4]) ) p_trend = result.pvalue# Adjust P value for one-sided testsif (p_tails ==1) & (p_trend !=1): p_trend = p_trend /2if result.statistic >0else1- p_trend /2 resp_pv[i] = p_trend# Check if both the control and treatment groups have at least one observation in any of the response categoriesif (control_row.sum() !=0) and (treatment_row.sum() !=0):# Perform Fisher's exact test on the 2x2 table _, p_fisher_val = fisher_exact(rt2.loc[[treatment_denominator, treatment]])# Adjust P value for one-sided testsif (p_tails ==1) & (p_fisher_val !=1): odds_ratio, _ = fisher_exact(rt2.loc[[treatment_denominator, treatment]], alternative='greater') p_fisher_val = p_fisher_val /2if odds_ratio >1else1- p_fisher_val /2 p_fisher[i] = p_fisher_val# Create DataFrame for results res = pd.DataFrame({'contrast': [f"{treatments_numerator[i]} vs. {treatment_denominator}"for i inrange(len(treatments_numerator))],"time_window": f"{day_ini}-{day_fin}",'p.trend': resp_pv,'p.fisher': p_fisher })return response, res
Contingency table (quaternary/4-level response):
response pd sd pr cr
group
control 10 5 0 0
therapy_a 4 11 0 0
therapy_b 9 6 0 0
therapy_combo_ab 2 10 3 0
Contingency table (binary response):
responder False True
group
control 10 5
therapy_a 4 11
therapy_b 9 6
therapy_combo_ab 2 13
contrast time_window p.trend p.fisher
0 therapy_a vs. control 0-6 0.030860 0.065595
1 therapy_b vs. control 0-6 0.709521 1.000000
2 therapy_combo_ab vs. control 0-6 0.002044 0.007775
While both Fisher’s exact test and the chi-squared test assess the association between categorical variables,
they have different underlying assumptions and calculations, which can lead to different P values even for the
same 2x2 contingency table.
Fisher’s exact test
Exact Calculation: calculates the exact probability of observing the given contingency table
(or something more extreme) under the null hypothesis of no association.
Assumptions: assumes that the row and column totals in the contingency table are fixed.
Best for: small sample sizes or sparse tables where the expected cell counts are low.
More computationally intensive: can be computationally expensive for large tables.
Chi-squared test
Approximation: uses the chi-squared distribution to approximate the P value.
Assumptions: assumes that the expected cell frequencies in each cell are sufficiently large (usually
at least 5) for the chi-squared approximation to be valid.
Best for: larger sample sizes and tables where the expected cell counts are not too low.
Less computationally intensive: generally faster to compute than Fisher’s exact test.
Code
import pingouin as pgpg.chi2_independence( response_table[response_table['group'].isin(['control', 'therapy_a'])], x='group', y='responder',# correction=True,)
c:\Users\Sébastien\Documents\data_science\biostatistics\Bonato_tutorial_python\.env\Lib\site-packages\pingouin\contingency.py:151: UserWarning: Low count on observed frequencies.
warnings.warn(f"Low count on {name} frequencies.")
We might see different P values between Fisher’s exact and chi-squared tests, for multiple reasons:
Small sample size: if the sample size is small, Fisher’s exact test will generally give a more
accurate P value, while the chi-squared test might be less reliable due to the approximation.
Sparse tables: if the contingency table has cells with very low expected counts, the chi-squared
approximation might not be accurate, leading to different P values compared to Fisher’s exact test.
Yates’ continuity correction: some implementations of the chi-squared test apply Yates’ continuity
correction for 2x2 tables. This correction can make the chi-squared test more conservative (resulting in
higher P values).
While both coin::independence_test with distribution = 'exact' and
statsmodels.stats.contingency_tables.test_ordinal_association (or coin::lbl_test
linear-by-linear association test) perform the Cochran-Armitage trend test, they differ in their approach. The
R function uses an exact permutation test, making no assumptions about the data distribution and
often yielding results closer to Fisher’s exact test, particularly beneficial for small sample sizes common in
TGI studies. In contrast, the Python function relies on the chi-squared asymptotic distribution,
which may be less accurate for smaller or sparse datasets.
Sparse data (e.g., zero counts in some response categories) can affect the accuracy of the chi-squared
approximation used to calculate the P value. For instance, the asymptotic test might suggest a
significant trend (P=0.031) for ‘therapy_a’ vs. ‘control’, while the exact permutation test, which is
more accurate for sparse data, might not confirm this significance (P=0.066, as obtained in R). While a trend
might appear visually, the exact test provides a more reliable assessment, especially with sparse data or
small sample sizes.
This discrepancy arises because zero counts reduce the effective sample size and can invalidate the
chi-squared approximation. In more extreme cases, if the contingency table lacks enough non-zero cells to
assess a trend, the Cochran-Armitage test cannot be performed, resulting in NaN values for the P
value.
Code
rt = response_table[response_table['group'].isin(['control', 'therapy_combo_ab'])]print( Table( pd.crosstab( rt['group'], rt['response'], dropna=False# Include all categories even with zero counts ), shift_zeros=False ).test_ordinal_association(# col_scores=np.array([1, 2, 3, 4]) ))
The Cochran-Armitage test for trend can also examine whether there’s a consistent directional shift in the
proportions of observations across ordered categories. In the current TGI study, these categories represent
the severity of tumor response, ranging from ‘pd’ (progressive disease) to ‘cr’ (complete response). The test
assigns scores to these categories, reflecting their inherent order (‘pd’ < ‘sd’ < ‘pr’ < ‘cr’).
A positive Z-value, such as the 3.0837 observed for ‘therapy_combo_ab’ vs. ‘control’, signifies a
positive trend. This means that as we move from the control group (row_score=0, default) to the
‘therapy_combo_ab’ group (row_score=1), there’s a shift towards the categories with higher
scores. In practical terms, this indicates that the combination therapy is associated with a greater
proportion of animals experiencing better responses (‘pr’ or ‘cr’) and a lower proportion experiencing ‘pd’,
the least desirable outcome.
This positive trend aligns with the expected therapeutic effect, where the combination therapy is
hypothesized to be more effective than the control in inducing tumor regression. The magnitude of the Z-value
(3.0837) further suggests that this trend is quite strong, indicating a substantial difference in the
distribution of response categories between the two groups.
4.5 Analysis of synergistic effects
The quest for effective cancer treatment hinges on understanding and exploiting synergy between drugs. This
arises from the inherent heterogeneity of tumors, where diverse subclones limit the efficacy of single-agent
therapies. By combining treatments with distinct targets, we aim to overcome this challenge and enhance
therapeutic impact.
Two key definitions of synergy exist in oncology:
Exceeding the effect of the most potent single agent, referred to as the highest single agent or
HSA effect, or
Exceeding the sum of individual effects, referred to as independent “Bliss” synergy.
Aligning the statistical methodology with the chosen synergy definition is crucial for accurate assessment
and interpretation.
4.5.1
HSA
Evaluating therapeutic synergy often involves statistical tests similar to those used for comparing
single-agent treatments to controls. However, in this context, the focus shifts to comparing the combination
therapy to the most effective single agent (HSA - Highest Single Agent). Since these comparisons
typically occur within the same animal study, we can assess synergy by testing the null hypothesis that the
combination therapy (AB) is not superior to the best single agent (A or B). This can be represented
by the hypothesis: \(H_0: \beta_{AB} < \max
\left(\beta_A,\beta_B\right)\), where \(\beta\) represents the
treatment effect (e.g., on tumor volume or growth rate).
4.5.2 Bliss
4.5.2.1 Bliss independence synergy
Bliss independence synergy assesses whether the combined effect of two drugs exceeds the expected
additive effect if they acted independently. It’s based on the idea that the probability of a tumor cell
surviving both drugs \(S_{AB}\) is the product of the individual survival
probabilities \(S_A\) and \(S_B\), such
as: \(P(S_A \cap S_B) = P(S_A) \ast P(S_B)\) if the drugs act
independently, as explained by Demidenko
and colleauges. Synergy occurs when \(P(S_A \cap S_B) < P(S_A) \ast
P(S_B)\), indicating a greater-than-additive effect.
In terms of tumor volumes, this translates to comparing the log-transformed tumor volumes of the
combination therapy \(Y_{AB}\) to the sum of the log-transformed tumor
volumes of the single agents \(Y_A\) and \(Y_B\), all relative to the control group \(Y_C\). Since \(P(S_A \cap S_B) = Y_{AB} /
Y_C\), \(P(S_A) = Y_A / Y_C\) and \(P(S_B) = Y_B / Y_C\), the null hypothesis for Bliss independence synergy
is:
This equation tests whether the combined effect (on the log scale) is simply the sum of the individual
effects or if there’s a synergistic interaction.
4.5.2.2 Visualization
We can visualize tumor growth curves for different treatment groups, including a control group, two
single-agent groups, a combination therapy group, and a theoretical “Bliss” curve representing the
expected outcome if the single agents acted independently and additively.
Code
def plot_bliss( dataset: pd.DataFrame, single_1_name: str, single_2_name: str, combo_name: str, control_name: str='control', ln_scale: bool=False, backend_seaborn: bool=True, colormap: str="viridis") ->None:"""Plots tumor growth curves with 95% confidence intervals and Bliss independence line. Args: dataset (pd.DataFrame): DataFrame with columns 'day', 'group', 'animal', and 'tv' (tumor volume). single_1_name (str): Name of the first single-agent treatment group. single_2_name (str): Name of the second single-agent treatment group. combo_name (str): Name of the combination treatment group. control_name (str, optional): Name of the control group. Defaults to 'control'. ln_scale (bool, optional): If True, plot on natural log scale. Defaults to False. backend_seaborn (bool, optional): If True, plots with Seaborn, else uses Matplotlib. colormap (str, optional): Colormap to use for group colors. Defaults to "viridis". """# Filter data for relevant groups included_groups = [control_name, single_1_name, single_2_name, combo_name] dataset_list = dataset[dataset['group'].isin(included_groups)].copy()# Calculate summary statistics for each group and day summ_stats = summary_stats_tgi(dataset_list)# Calculate Bliss independence valuesdef bliss_values(x: pd.DataFrame) -> pd.DataFrame: new_x = pd.DataFrame({'day': [x['day'].unique()[0]],'group': ['Bliss'],'est': [ x[x['group'] == single_1_name]['est'].values[0] * x[x['group'] == single_2_name]['est'].values[0] / x[x['group'] == control_name]['est'].values[0] ] })return pd.concat([x, new_x], ignore_index=True) summ_stats = ( summ_stats .groupby('day') .apply(bliss_values) .reset_index(drop=True) )# Prepare for plotting groups = [control_name, single_1_name, single_2_name, combo_name, 'Bliss'] summ_stats['group'] = pd.Categorical(summ_stats['group'], categories=groups)# Plotif backend_seaborn: ax = sns.lineplot( data=TGI_dataset, x='day', y='tv', hue='group', style='group', dashes=False, markers=["o", "s", "^", "D"], estimator=gmean, errorbar=('ci', 95), n_boot=100, palette='viridis', err_style='band', lw=2, markersize=8) ax.set_xticks(TGI_dataset['day'].unique()) ax.set_xticklabels(TGI_dataset['day'].unique(), fontsize=10) ax.set_xlabel("Days", fontsize=12) ax.set_ylabel("Tumor volume (mm³)", fontsize=12) ax.set_title("Tumor growth curves", fontsize=14) ax.grid(which='major', axis='y', linestyle='--')if ln_scale: ax.set_yscale("log")else: plot_tgi_stats( summ_stats[summ_stats['group'] !='Bliss'], # Plot only the non-Bliss groups ln_scale=ln_scale, colormap=colormap)# Add Bliss line bliss_data = summ_stats[summ_stats['group'] =='Bliss'] plt.plot( bliss_data['day'], bliss_data['est'], color='red', linestyle='dashed', linewidth=2, label='Bliss')# Show plot plt.legend(loc='upper left', fontsize=10);
The Bliss independence model predicts the combined effect of two drugs assuming they act independently of
each other. In other words, it estimates the expected effect if the drugs simply add their individual
effects together. Synergy occurs when the combined effect of the drugs is greater than what would be
expected if they acted independently.
The dashed red Bliss line on the plot represents this predicted combined effect under the assumption of
independence. Since the Bliss line is above the combination therapy curve, it indicates that the
combination therapy is performing better than what the Bliss independence model would predict. This
suggests that the two drugs are working together in a synergistic way to inhibit tumor growth more
effectively than simply adding their individual effects.
4.5.2.3 Inference
Let’s define a statistical test for Bliss independence synergy, which checks if the combined effect of
two drugs is greater than the sum of their individual effects. The underlying assumption (null hypothesis)
is that the drugs act independently, meaning their combined effect is simply additive.
The test statistic (\(S\)) is a modified t-statistic derived from the
principles of linear models and the properties of the t-distribution. It’s based on the average
log-transformed tumor volumes (\({\overline{y}}_{ij}^{\ast }\)) of the
different groups (combination therapy AB, single agents A and B, and control C). This statistic
essentially measures the difference between the combined effect and the sum of individual effects:
In the numerator, we recognize the term that essentially calculates the difference between the combined
effect (AB) and the sum of the individual effects (A and B), all relative to the control (C). This term is
scaled by a factor that adjusts for the degrees of freedom \(\sum_i{n_i} -
4\) in the t-distribution. Finally, the denominator estimates the standard error of this
difference.
Bliss synergy, i.e., \(\exp({\overline{y}}_{AB}^{\ast }+{\overline{y}}_C^{\ast
}-{\overline{y}}_A^{\ast }-{\overline{y}}_B^{\ast})\), is statistically significant if it is
lower than 1 (meaning the combined effect is greater than the sum of individual effects)
and exceeds the one-sided critical value of the respective t-distribution. This indicates
that the combined effect is significantly greater than expected from simply adding the individual effects.
Code
from patsy.contrasts import Treatmentfrom statsmodels.stats.contrast import ContrastResultsdef bliss_synergy_test( dataset: pd.DataFrame, single_1_name: str, single_2_name: str, combo_name: str, time_points: List[int], control_name: str='control', baseline_adjusted: bool=False, baseline: int=0,) -> pd.DataFrame:""" Tests for Bliss independence synergy between two single-agent treatments and their combination. Args: dataset (pd.DataFrame): TGI dataset with columns 'day', 'group', 'animal', and 'tv' (tumor volume). single_1_name (str): Name of the first single-agent treatment group. single_2_name (str): Name of the second single-agent treatment group. combo_name (str): Name of the combination treatment group. time_points (List[int]): List of time points (days) to analyze. control_name (str, optional): Name of the control group. Defaults to 'control'. baseline_adjusted (bool, optional): Whether to adjust for baseline tumor volume. Defaults to False. baseline (int, optional): Baseline day for adjustment. Required if `baseline_adjusted` is True. Defaults to None. Returns: pd.DataFrame: A DataFrame with the following columns: - contrast: The comparison being made (e.g., "treatment_combo_ab vs. Bliss"). - day: The time point (day) of the analysis. - synergy: exp(estimate), where 'estimate' is from the contrast. - lb95_synergy: Lower bound of the 95% confidence interval for synergy. - ub95_synergy: Upper bound of the 95% confidence interval for synergy. - pvalue: One-sided P value from the t-distribution. - df: Degrees of freedom. - baseline_adj: Whether the estimate was adjusted for baseline day. """# Filter data for relevant groups and time points (including baseline if adjusting) relevant_days = time_points +\ [baseline] if baseline_adjusted else time_points groups = [control_name, single_1_name, single_2_name, combo_name] tv_data = dataset[ dataset['group'].isin(groups) & dataset['day'].isin(relevant_days) ].copy()# Log-transform tumor volumes tv_data['log_tv'] = np.log(tv_data['tv'])# Baseline adjustment (if applicable)if baseline_adjusted: tv_data['log_tv_baseline'] = ( tv_data .groupby('animal') .log_tv .transform(lambda x: x[tv_data['day'] == baseline].values[0]) ) # Calculate adjusted log_tv (difference from baseline) tv_data['log_tv_adjusted'] = tv_data['log_tv'] - tv_data['log_tv_baseline']# Remove the baseline day tv_data = tv_data[tv_data['day'] != baseline]# Convert 'group' and 'day' to categorical variables with appropriate ordering tv_data['group'] = pd.Categorical(tv_data['group'], categories=groups, ordered=True) day_idx =sorted(tv_data['day'].unique()) tv_data['day'] = pd.Categorical( tv_data['day'], categories=day_idx, ordered=True)# Perform analysis for each time point model_estimates = [] response_variable ='log_tv'ifnot baseline_adjusted else'log_tv_adjusted'for day in day_idx:# Filter data for the current time point tv_data_day = tv_data[tv_data['day'] == day]# Group means (using the appropriate response variable based on baseline_adjusted) group_means = tv_data_day.groupby('group')[response_variable].mean() y_AB = group_means[combo_name] y_C = group_means[control_name] y_A = group_means[single_1_name] y_B = group_means[single_2_name]# Group sizes n_i = tv_data_day['group'].value_counts()# Calculate numerator numerator = (y_AB + y_C - y_A - y_B) * np.sqrt(np.sum(n_i) -4)# Calculate denominator sum_sq_diff =0for group, mean in group_means.items(): group_data = tv_data_day[tv_data_day['group'] == group][response_variable] sum_sq_diff += np.sum((group_data - mean)**2) denominator = np.sqrt(sum_sq_diff * np.sum(1/ n_i))# Calculate t-statistic t_stat = numerator / denominator# Degrees of freedom df = np.sum(n_i) -4# Calculate P value (one-sided) p_value = t_dist.cdf(t_stat, df)# Calculate synergy estimate estimate = y_AB + y_C - y_A - y_B# Calculate confidence intervals alpha =0.1# For 90% confidence intervals t_critical = t_dist.ppf(1- alpha /2, df) se = (y_AB + y_C - y_A - y_B) / t_stat # Standard error lb95_synergy = np.exp((estimate) - t_critical * se) ub95_synergy = np.exp((estimate) + t_critical * se)# Store results model_estimates.append({'contrast': f'{combo_name} vs. Bliss','day': day,'synergy': np.exp(estimate),'lb95_synergy': lb95_synergy,'ub95_synergy': ub95_synergy,'pvalue': p_value,'df': df,'baseline_adj': baseline_adjusted, })return pd.DataFrame(model_estimates)
# Generate another TGI_dataset with a substantial synergynp.random.seed(12345)# Simulate the vehicle (control) group datavehicle_group = simulate_exp_growth( group_name="control", duration=18, initial_tumor_volume=100, group_growth_rate_day_pct=5, growth_rate_cv=0.1, residual_cv=0.25, n_animals=15, max_tumor_volume=2000)# Simulate data for the single-agent therapy A grouptherapy_a_group = simulate_exp_growth( group_name="therapy_a", duration=18, initial_tumor_volume=100, group_growth_rate_day_pct=4, growth_rate_cv=0.1, residual_cv=0.25, n_animals=15, max_tumor_volume=2000)# Simulate data for the single-agent therapy B grouptherapy_b_group = simulate_exp_growth( group_name="therapy_b", duration=18, initial_tumor_volume=100, group_growth_rate_day_pct=4, growth_rate_cv=0.1, residual_cv=0.25, n_animals=15, max_tumor_volume=2000)# Simulate data for the combination therapy A+B grouptherapy_ab_group = simulate_exp_growth( group_name="therapy_combo_ab", duration=18, initial_tumor_volume=100, group_growth_rate_day_pct=1.1, growth_rate_cv=0.1, residual_cv=0.25, n_animals=15, max_tumor_volume=2000)# Combine all group data into a single DataFrameTGI_dataset_12345 = pd.concat( [ vehicle_group, therapy_a_group, therapy_b_group, therapy_ab_group ], ignore_index=True)
Note that compared to the case where we adjust for the baseline without using a linear model, the linear
model approach lm(lnTV ~ lnTV.baseline + Group, data = TV.data.day) uses up one extra
degree of freedom due to the estimation of the baseline coefficient. This reduction in degrees of
freedom can have a slight impact on the p-values and confidence intervals, especially when the sample size
is small. However, in most cases, the effect is minimal.
4.6 Powering studies
4.6.1 Statistical power
Statistical power is the probability of detecting
a true treatment effect when it actually exists. In simpler terms, it’s the chance that your study
will correctly identify a difference between a treatment and a control group if a real difference exists.
TGI studies often exhibit high variability due to factors like:
Choice of endpoint: different endpoints (tumor volume, growth rate, survival time) have varying levels
of variability.
Measurement techniques: measurement errors can introduce variability.
Biological variability: inherent differences between animals and tumor models contribute to variability.
Achieving high statistical power is crucial for reliable results. It depends on several factors:
Sample size: larger sample sizes generally lead to higher power, as they provide more data to detect a
true effect.
Effect size: the larger the true difference between the treatment and control groups, the easier it is
to detect, leading to higher power.
Variability: lower variability within and between groups makes it easier to distinguish a treatment
effect, increasing power.
Significance level (alpha): The significance level (typically 0.05) is the threshold for determining
statistical significance. A higher alpha (e.g., 0.10) increases the chance of detecting an effect (higher
power) but also increases the risk of a false positive (Type I error).
Beyond increasing sample size, several strategies can enhance power in TGI studies:
Model selection: choose animal and tumor models with low intra- and inter-model variability.
Engraftment failures: start with extra animals to account for potential engraftment failures, ensuring
sufficient sample size.
Measurement error: implement good practices to minimize measurement error (e.g., standardized
techniques, blinded measurements).
Longitudinal analysis: analyze data over time (longitudinally) whenever possible, as it provides more
information and increases power compared to single time-point analyses.
Baseline adjustment: for single time-point analyses, adjust for baseline tumor size to account for
initial variability.
Covariate modeling: carefully consider how covariates (e.g., dose, weight) are included in the analysis.
Treat dose as a continuous variable if appropriate, and avoid unnecessary categorization.
4.6.2 Calculating sample size for two-sample t-tests
Calculating the sample size needed for a study to reliably detect a true treatment effect (statistical
power) involves several considerations. A common target power level is 80% (\(1 -
\beta = 0.8\), where \(\beta\) is the probability of a false
negative). The conventional cutoff for a false positive (Type I error) is \(\alpha
= 0.05\). After optimizing the study design and analysis approach, we can determine the necessary
sample size to achieve the desired power and minimize the risk of both false positive and false negative
results.
To determine the necessary sample size \(n\) for a two-sample t-test, we
need to consider:
Desired power (\(1 - \beta\)): the probability of detecting a true
effect (commonly 80%).
Significance level (\(\alpha\)): the probability of a false positive
(typically 0.05).
Effect size (\(\theta\)): the expected difference between groups (e.g.,
a 50% reduction in tumor volume).
Intra-group variances (\(\sigma_1^2\) and \(\sigma_2^2\)): the variability within each group.
The sample size is calculated by solving the equation \(\text{Power} = P[t(\nu,
\delta) \ge t_{1 - \alpha; \nu}]\), where:
\(t(\nu, \delta)\) is a non-central
t-distribution with degrees of freedom \(\nu = n - 2\) (with \(n = n_1 + n_2\) the sum of animals per group) degrees of freedom.
\(\delta\) is the non-centrality parameter calculated based on the
effect size \(\theta\) and the intra-group variances, i.e., \(\delta = \frac{\log(1 + \theta)}{\sigma}\), and \(\sigma =\sqrt{\frac{\sigma_1^2}{n_1}+\frac{\sigma_2^2}{n_2}}\), as shown in
the Welch’s
t-test.
\({t}_{1 - \alpha;\nu }\) is the critical value (\(1 - \alpha\) th quantile) of the central t-distribution for the given
significance level (\(\alpha\)) and degrees of freedom (\(\nu\)).
If the linear model includes additional parameters (e.g., baseline adjustment), the degrees of freedom in
the equation are adjusted accordingly (e.g., \(\nu = n - 3\) to account for
one additional parameter to be estimated).
The plots below illustrate the core concept of statistical power in hypothesis testing. Specifically, we
want to visualize:
The relationship between Type II error (\(\beta\)) and power (\(1-\beta\)): power
is the probability of correctly rejecting a false null hypothesis. Beta (\(\beta\)) is the probability of failing to reject a false null
hypothesis (a Type II error). They are complementary: higher power means lower beta, and vice versa.
How effect size (\(\delta\)) influences power: a larger effect size (a
bigger difference between groups, for example) makes it easier to detect, leading to higher power.
The role of the critical value: this value, derived from the chosen significance level (\(\alpha\)), sets the threshold for rejecting the null hypothesis.
Code
from scipy.stats import t, nctfrom scipy.optimize import fsolve# Parametersdf =10# Degrees of freedomalpha =0.05# Significance level# Calculate critical valuecritical_value = t.ppf(1- alpha, df)# Generate x-values for plottingx = np.linspace(t.ppf(0.001, df), t.ppf(0.999, df), 100)# Generate y-values for central t-distributiony_central = t.pdf(x, df)# Function to calculate powerdef calculate_power(delta): y_noncentral = nct.pdf(x, df, delta) power = y_noncentral[x > critical_value].sum() / y_noncentral.sum() # Approximate powerreturn power - target_power # Adjust for fsolve# Target power levels (which correspond to 1-beta)target_powers = [0.2, 0.5, 0.8]# Create subplotsfig, axes = plt.subplots(1, 3, figsize=(15, 5))for i, target_power inenumerate(target_powers):# Find delta for target power delta_optimized = fsolve(calculate_power, 2) # Start with an initial guess of 2# Generate y-values for non-central t-distribution with optimized delta y_noncentral_opt = nct.pdf(x, df, delta_optimized)# Plot the distributions axes[i].plot(x, y_central, label='Central t') axes[i].plot(x, y_noncentral_opt, label='Non-central t')# Shade the rejection region axes[i].fill_between(x[x > critical_value], 0, y_central[x > critical_value], alpha=0.5, color='mediumseagreen', label='Rejection Region')# Shade the beta area axes[i].fill_between(x[x < critical_value], 0, y_noncentral_opt[x < critical_value], alpha=0.5, color='orange', label='Beta')# Add vertical line for critical value axes[i].axvline(x=critical_value, color='black', linestyle='--', label='Critical value')# Add labels and legend axes[i].set_title(fr"$\beta=$ {1-target_power:.0%} | $\delta=$ {delta_optimized[0]:.2f}") axes[i].set_xlabel('t-value') axes[i].set_ylabel('Probability density') axes[i].legend()# Adjust layoutplt.tight_layout();
How It Works:
Two distributions: we plot a central t-distribution (representing the null hypothesis) and a
non-central t-distribution (representing the alternative hypothesis when an effect exists).
Shifting with effect size: the non-central t-distribution shifts further from the center as the effect
size (\(\delta\)) increases.
Critical value as a boundary: the critical value on the central t-distribution marks the rejection
region.
Beta (\(\beta\)) and Power (\(1-\beta\)): the area under the non-central t-distribution to the
left of the critical value represents beta (\(\beta\)), the
probability of a Type II error. The area to the right represents power (\(1-\beta\)).
The critical value is implicitly considered within the power calculation. Statistical software, online
calculators, or the custom functions defined below, use the chosen alpha to determine the critical value
“behind the scenes.” They then use this critical value, along with the desired power and effect size, to
calculate the required sample size.
4.6.3 Calculating sample size for ANOVA and ANCOVA
When comparing more than two groups (e.g., three different treatment groups), the standard power
calculation for a t-test needs modification:
Degrees of freedom (\(\nu\)): the formula for degrees of freedom
changes to account for the multiple groups and any additional factors in the model: \(\nu = (n - 1) * j - 2 - z\), where:
\(n\) is the number of subjects per group.
\(j\) is the number of groups.
\(z\) is the number of covariates (e.g., in ANCOVA)
Standard deviation (\(\sigma\)): the way we calculate the standard
deviation also changes:\(\sigma = \sqrt{2 * \sigma^2 / n}\), where
\(\sigma^2\) is the residual variance (variability within groups)
Using the correct formulas for degrees of freedom and standard deviation ensures we get a reliable estimate
of the sample size needed for the study. These adjustments consider the increased complexity of the analysis
with multiple groups and potential covariates.
Code
from scipy.optimize import brentqdef pwr_gt2_grps(n, sigma, eff, two_tailed, n_groups, alpha, method):""" Calculates the power for more than two groups. Args: n: Sample size per group. sigma: Residual standard deviation. eff: Effect size. two_tailed: Two-sided or one-sided test. n_groups: Number of groups. alpha: Significance level. method: "ANOVA" or "ANCOVA". Returns: Power of the test. """ df_n = (n -1) * n_groups - (method =="ANCOVA") se_n = (2/ n)**0.5* sigmaif two_tailed: alpha /=2 tcrit = t.ppf(1- alpha, df_n) noncen = np.log(1+ eff) / se_nreturn1- nct.cdf(tcrit, df=df_n, nc=noncen) + nct.cdf(-tcrit, df=df_n, nc=noncen)def pwr_2_grps(n1, n2, sigma1, sigma2, eff, two_tailed, alpha, method):""" Calculates the power for two groups. Args: n1: Sample size for group 1. n2: Sample size for group 2. sigma1: Standard deviation for group 1. sigma2: Standard deviation for group 2. eff: Effect size. two_tailed: Two-sided or one-sided test. alpha: Significance level. method: "ANOVA" or "ANCOVA". Returns: Power of the test. """ df_n = n1 + n2 -2- (method =="ANCOVA") se_n = (1/ n1 * sigma1**2+1/ n2 * sigma2**2)**0.5 alpha = alpha /2if two_tailed else alpha tcrit = t.ppf(1- alpha, df_n) noncen = np.log(1+ eff) / se_nreturn1- nct.cdf(tcrit, df=df_n, nc=noncen) + nct.cdf(-tcrit, df=df_n, nc=noncen)def power_function( sigma: float| List[float] =.25, n: float| List[float] =None, eff: float=0.5, power: float=0.8, n_groups=2, alpha=0.05, two_tailed=True, method="ANCOVA") -> pd.DataFrame:""" Calculates power or other parameters for a given set of inputs. Args: sigma: Residual standard deviation (or list for two groups). n: Sample size per group (or list for two groups). eff: Effect size. power: Desired power. n_groups: Number of group. alpha: Significance level. two_tailed: Two-sided test or one-sided (Defaults to True). method: "ANOVA" or "ANCOVA". Returns: A pandas DataFrame with calculated parameters. """ pars = [ sigma isNone, n isNone, eff isNone, power isNone]ifsum(pars) !=1:return"Error: one and only one parameter should be NULL" par = pars.index(True)if par ==3and n_groups >2: # Calculate 'power' for more than 2 groups df_n = (n -1) * n_groups - (method =="ANCOVA") se_n = (2/ n)**0.5* sigmapow= pwr_gt2_grps( n=n, sigma=sigma, eff=eff, two_tailed=two_tailed, n_groups=n_groups, alpha=alpha, method=method)return pd.DataFrame({"sigma": [sigma],"n_groups": [n_groups],"n": [n],"df": [df_n],"se": [se_n],"eff": [eff],"power": [pow],})elif par ==3and n_groups ==2: # Calculate 'power' for two groups n1, n2 = n[0], n[1] ifisinstance(n, list) else n, n s1, s2 = sigma[0], sigma[1] ifisinstance(sigma, list) else sigma, sigma df_n = (n1 + n2 -2) - (method =="ANCOVA") se_n = (s1**2/ n1 + s2**2/ n2)**0.5pow= pwr_2_grps( n1=n1, n2=n2, sigma1=s1, sigma2=s2, eff=eff, two_tailed=two_tailed, alpha=alpha, method=method)return pd.DataFrame({"sigma1": [s1],"sigma2": [s2],"n1": [n1],"n2": [n2],"df": [df_n],"se": [se_n],"eff": [eff],"power": [pow],})elif par !=3and n_groups >2: # Calculate other parametersif sigma isNone: f =lambda sc: power - pwr_gt2_grps( n=n, sigma=sc, eff=eff, two_tailed=two_tailed, n_groups=n_groups, alpha=alpha, method=method) sc = brentq(f, 0.01, 20) # looking up at values of sigma between 0.01 and 20 df_n = (n -1) * n_groups - (method =="ANCOVA") se_n = (2/ n)**0.5* screturn pd.DataFrame({"sigma": [sc],"n_groups": [n_groups],"n": [n],"df": [df_n],"se": [se_n],"eff": [eff],"power": [power],})elif n isNone: f =lambda ns: power - pwr_gt2_grps( n=ns, sigma=sigma, eff=eff, two_tailed=two_tailed, n_groups=n_groups, alpha=alpha, method=method) ns = brentq(f, 2, 400) # looking up at values of n between 2 and 400 df_n = (ns -1) * n_groups - (method =="ANCOVA") se_n = (2/ ns)**0.5* sigmareturn pd.DataFrame({"sigma": [sigma],"n_groups": [n_groups],"n": [ns],"df": [df_n],"se": [se_n],"eff": [eff],"power": [power],})else: # eff is None: f =lambda ef: power - pwr_gt2_grps( n=n, sigma=sigma, eff=ef, two_tailed=two_tailed, n_groups=n_groups, alpha=alpha, method=method) ef = brentq(f, .1, 10) # looking up at values of eff between 0.1 and 10 df_n = (n -1) * n_groups - (method =="ANCOVA") se_n = (2/ n)**0.5* sigmareturn pd.DataFrame({"sigma": [sigma],"n_groups": [n_groups],"n": [n],"df": [df_n],"se": [se_n],"eff": [ef],"power": [power],})elif par !=3and n_groups ==2: # Calculate other parameters 2 groupsif sigma isNone: n1, n2 = (n[0], n[1]) ifisinstance(n, list) else (n, n) f =lambda sc: power - pwr_2_grps( n1=n1, n2=n2, sigma1=sc, sigma2=sc, eff=eff, two_tailed=two_tailed, alpha=alpha, method=method) sc = brentq(f, 0.01, 20) # looking up at values of sigma between 0.01 and 20 df_n = (n1 + n2 -2) - (method =="ANCOVA") se_n = (sc**2/ n1 + sc**2/ n2)**0.5return pd.DataFrame({"sigma1": [sc],"sigma2": [sc],"n1": [n1],"n2": [n2],"df": [df_n],"se": [se_n],"eff": [eff],"power": [power],})elif n isNone: s1, s2 = (sigma[0], sigma[1]) ifisinstance(sigma, list) else (sigma, sigma) f =lambda ns: power - pwr_2_grps( n1=ns, n2=ns, sigma1=s1, sigma2=s2, eff=eff, two_tailed=two_tailed, alpha=alpha, method=method) ns = brentq(f, 2, 400) # looking up at values of n between 2 and 400 df_n = (ns + ns -2) - (method =="ANCOVA") se_n = (s1**2/ ns + s2**2/ ns)**0.5return pd.DataFrame({"sigma1": [s1],"sigma2": [s2],"n1": [ns],"n2": [ns],"df": [df_n],"se": [se_n],"eff": [eff],"power": [power],})else: # eff is None: n1, n2 = (n[0], n[1]) ifisinstance(n, list) else (n, n) s1, s2 = (sigma[0], sigma[1]) ifisinstance(sigma, list) else (sigma, sigma) f =lambda ef: power - pwr_2_grps( n1=n1, n2=n2, sigma1=s1, sigma2=s2, eff=ef, two_tailed=two_tailed, alpha=alpha, method=method) ef = brentq(f, .1, 10) # looking up at values of eff between 0.1 and 10 df_n = (n1 + n2 -2) - (method =="ANCOVA") se_n = (s1**2/ n1 + s2**2/ n2)**0.5return pd.DataFrame({"sigma1": [s1],"sigma2": [s2],"n1": [n1],"n2": [n2],"df": [df_n],"se": [se_n],"eff": [ef],"power": [power],})
While slight differences between scipy’s nct and R’s pt with ncp are
possible, they are usually minor and shouldn’t significantly affect the overall analysis in most cases.
However, it’s always good practice to be aware of these potential variations and take appropriate steps if
extremely high precision is required.
Moreover, when providing two distinct values for n1 and n2 (or s1
and s2), the solver returns the same value for both, effectively making n1 = n2 or
s1 = s2. This behavior is due to the limitations of the numerical solver brentq
being used, that is designed to find a single root of an equation within a given interval. In this case, the
equation represents the relationship between power, sample size, effect size, and standard deviation. If we
need to find finely tuned solutions, we may need to consider alternative optimization methods, e.g.,
constrained optimization, grid search, iterative approach or simulation, to explore the solution space more
effectively.
5
Conclusion
This tutorial serves as a comprehensive guide to tumor growth inhibition (TGI) studies, specifically designed
for researchers in the pharmaceutical industry tasked with prioritizing promising anticancer compounds. It
offers a deep dive into the critical aspects of preclinical animal studies, encompassing a wide range of
considerations from experimental design to data analysis.
The tutorial begins by exploring the foundational elements of TGI studies, including the selection of
appropriate animal models that accurately reflect human tumor biology and the identification of relevant
endpoints for assessing anticancer activity. It emphasizes the importance of choosing endpoints that not only
capture tumor growth dynamics but also reflect clinically meaningful outcomes, such as tumor volume, survival
time, and the expression of specific biomarkers.
Recognizing the inherent variability in biological systems, the tutorial addresses the crucial issue of
measurement errors, both random and non-random, and their potential impact on the interpretation of TGI data. It
provides strategies for minimizing these errors through careful experimental design, standardized procedures,
and the use of appropriate statistical methods. Moreover, it offers guidance on handling missing data, a common
challenge in preclinical studies, outlining various imputation techniques and their implications for data
analysis.
A core component of the tutorial focuses on statistical power and sample size determination. It elucidates the
factors influencing power, including the magnitude of the treatment effect, variability within the data, and the
chosen significance level. By providing a clear understanding of these concepts, the tutorial empowers
researchers to conduct power analysis and determine the minimum sample size required to reliably detect
meaningful differences between treatment groups.
Beyond experimental design, the tutorial delves into the statistical analysis of TGI data, covering a range of
methods tailored to different types of endpoints. It reviews techniques for analyzing continuous data, such as
tumor volume measurements, using t-tests, ANOVA, and regression analysis. It also explores survival analysis
methods, including Kaplan-Meier curves and Cox regression, for analyzing time-to-event data, such as time to
tumor progression or death. Additionally, it discusses methods for analyzing categorical endpoints, such as
tumor response rates, enabling researchers to comprehensively evaluate treatment efficacy.
Furthermore, the tutorial extends its scope to the analysis of drug combination synergy, a critical aspect of
anticancer drug development. It provides insights into evaluating the combined effects of multiple agents,
exploring concepts like additivity, synergy, and antagonism. By understanding these principles, researchers can
identify promising drug combinations that may enhance therapeutic outcomes.
In conclusion, this tutorial equips researchers with a comprehensive understanding of TGI studies, encompassing
experimental design, data analysis, and the assessment of drug combination synergy. It serves as a valuable
resource for navigating the complexities of preclinical drug development, ultimately contributing to the
identification and prioritization of effective anticancer therapies.
6 Session Information
Code
pip show lifelines
Name: lifelines
Version: 0.29.0
Summary: Survival analysis in Python, including Kaplan Meier, Nelson Aalen and regression
Home-page: https://github.com/CamDavidsonPilon/lifelines
Author: Cameron Davidson-Pilon
Author-email: cam.davidson.pilon@gmail.com
License: MIT
Location: c:\Users\Sébastien\Documents\data_science\biostatistics\Bonato_tutorial_python\.env\Lib\site-packages
Requires: autograd, autograd-gamma, formulaic, matplotlib, numpy, pandas, scipy
Required-by:
Note: you may need to restart the kernel to use updated packages.
Code
!python --versionprint("-------------")import pkg_resources# List of packages we want to check the versionpackages = ['numpy', 'pandas', 'matplotlib', 'seaborn', 'scipy', 'statsmodels', 'pingouin', 'lifelines']# Initialize an empty list to store the versionsversions = []# Loop over the packagesfor package in packages:try:# Get the version of the package version = pkg_resources.get_distribution(package).version# Append the version to the list versions.append(version)except pkg_resources.DistributionNotFound: versions.append('Not installed')# Print the versionsfor package, version inzip(packages, versions):print(f'{package}: {version}')